Trending in Quantitative Biology
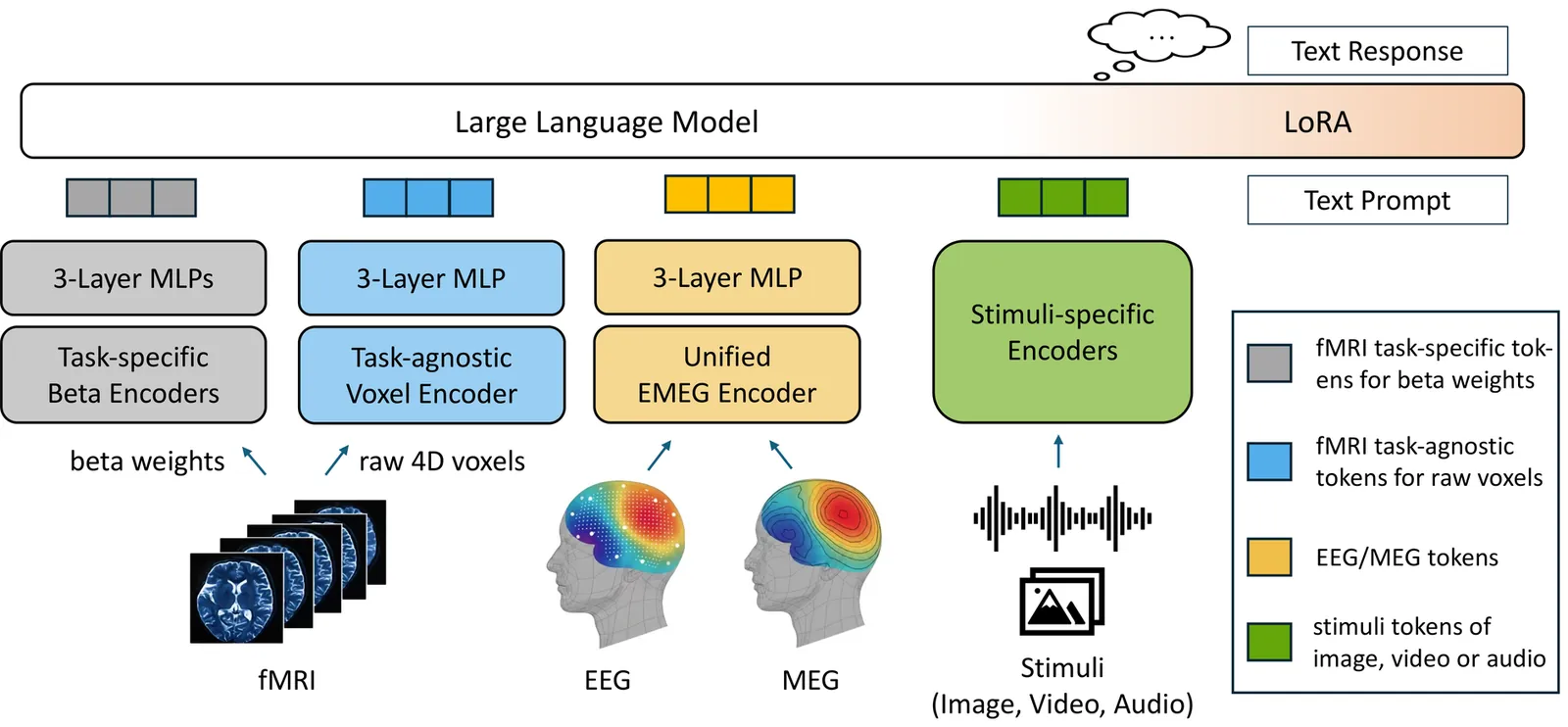
One Brain, Omni Modalities: Towards Unified Non-Invasive Brain Decoding with Large Language Models
Deciphering brain function through non-invasive recordings requires synthesizing complementary high-frequency electromagnetic (EEG/MEG) and low-frequency metabolic (fMRI) signals. However, despite their shared neural origins, extreme discrepancies have traditionally confined these modalities to isolated analysis pipelines, hindering a holistic interpretation of brain activity. To bridge this fragmentation, we introduce \textbf{NOBEL}, a \textbf{n}euro-\textbf{o}mni-modal \textbf{b}rain-\textbf{e}ncoding \textbf{l}arge language model (LLM) that unifies these heterogeneous signals within the LLM's semantic embedding space. Our architecture integrates a unified encoder for EEG and MEG with a novel dual-path strategy for fMRI, aligning non-invasive brain signals and external sensory stimuli into a shared token space, then leverages an LLM as a universal backbone. Extensive evaluations demonstrate that NOBEL serves as a robust generalist across standard single-modal tasks. We also show that the synergistic fusion of electromagnetic and metabolic signals yields higher decoding accuracy than unimodal baselines, validating the complementary nature of multiple neural modalities. Furthermore, NOBEL exhibits strong capabilities in stimulus-aware decoding, effectively interpreting visual semantics from multi-subject fMRI data on the NSD and HAD datasets while uniquely leveraging direct stimulus inputs to verify causal links between sensory signals and neural responses. NOBEL thus takes a step towards unifying non-invasive brain decoding, demonstrating the promising potential of omni-modal brain understanding.
2602.21522
Feb 2026Neurons and Cognition
Bootstrapping Life-Inspired Machine Intelligence: The Biological Route from Chemistry to Cognition and Creativity
Achieving advanced machine intelligence remains a central challenge in AI research, often approached through scaling neural architectures and generative models. However, biological systems offer a broader repertoire of strategies for adaptive, goal-directed behavior - strategies that emerged long before nervous systems evolved. This paper advocates a genuinely life-inspired approach to machine intelligence, drawing on principles from biology that enable robustness, autonomy, and open-ended problem-solving across scales. We frame intelligence as flexible problem-solving, following William James, and develop the concept of "cognitive light cones" to characterize the continuum of intelligence in living systems and machines. We argue that biological evolution has discovered a scalable recipe for intelligence - and the progressive expansion of organisms' "cognitive light cone", predictive and control capacities. To explain how this is possible, we distill five design principles - multiscale autonomy, growth through self-assemblage of active components, continuous reconstruction of capabilities, exploitation of physical and embodied constraints, and pervasive signaling enabling self-organization and top-down control from goals - that underpin life's ability to navigate creatively diverse problem spaces. We discuss how these principles contrast with current AI paradigms and outline pathways for integrating them into future autonomous, embodied, and resilient artificial systems.
2602.08079
Feb 2026Neurons and Cognition
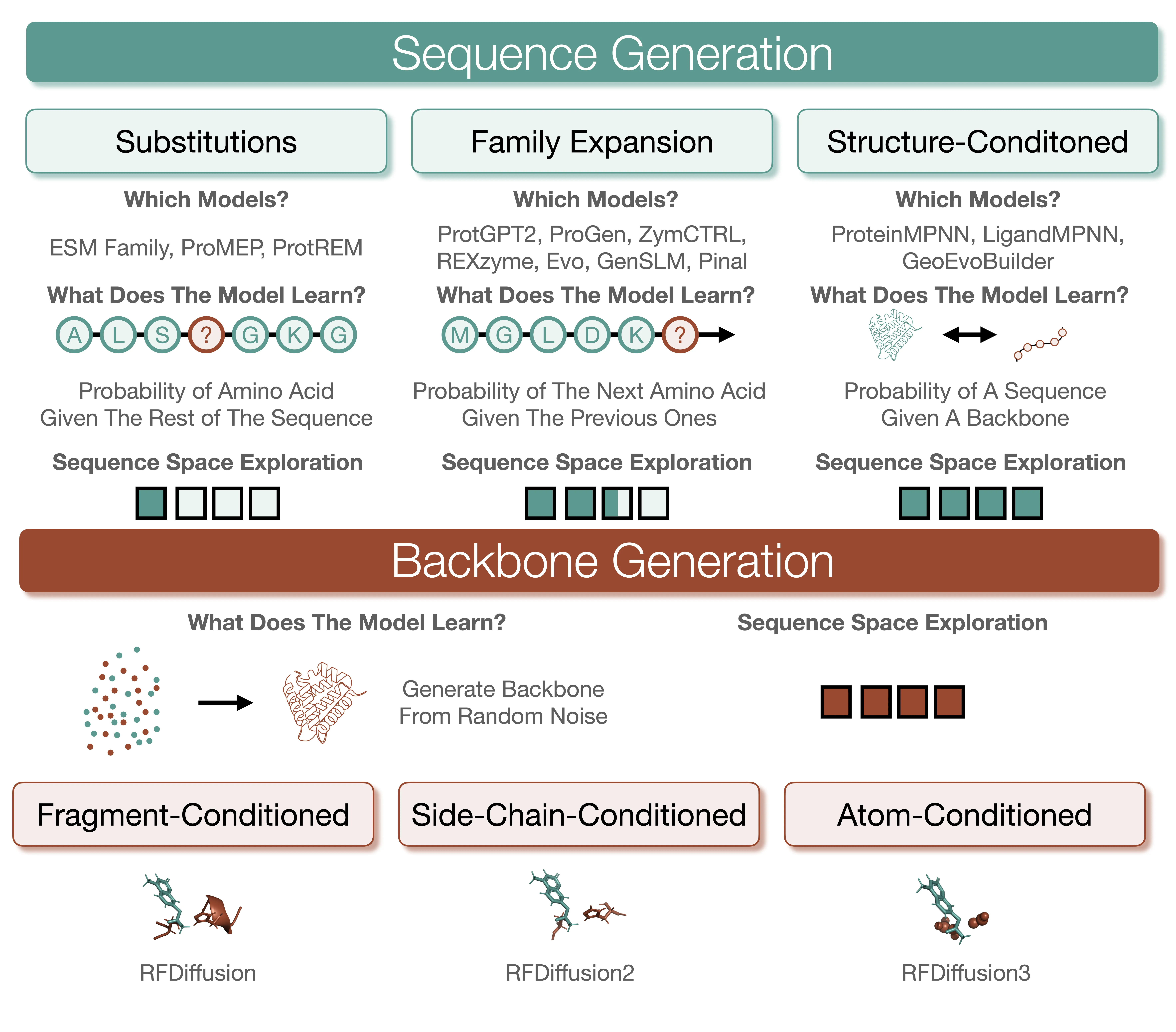
Generative AI for Enzyme Design and Biocatalysis
Sparked by innovations in generative artificial intelligence (AI), the field of protein design has undergone a paradigm shift with an explosion of new models for optimizing existing enzymes or creating them from scratch. After more than one decade of low success rates for computationally designed enzymes, generative AI models are now frequently used for designing proficient enzymes. Here, we provide a comprehensive overview and classification of generative AI models for enzyme design, highlighting models with experimental validation relevant to real-world settings and outlining their respective limitations. We argue that generative AI models now have the maturity to create and optimize enzymes for industrial applications. Wider adoption of generative AI models with experimental feedback loops can speed up the development of biocatalysts and serve as a community assessment to inform the next generation of models.
2602.03779
Feb 2026Biomolecules
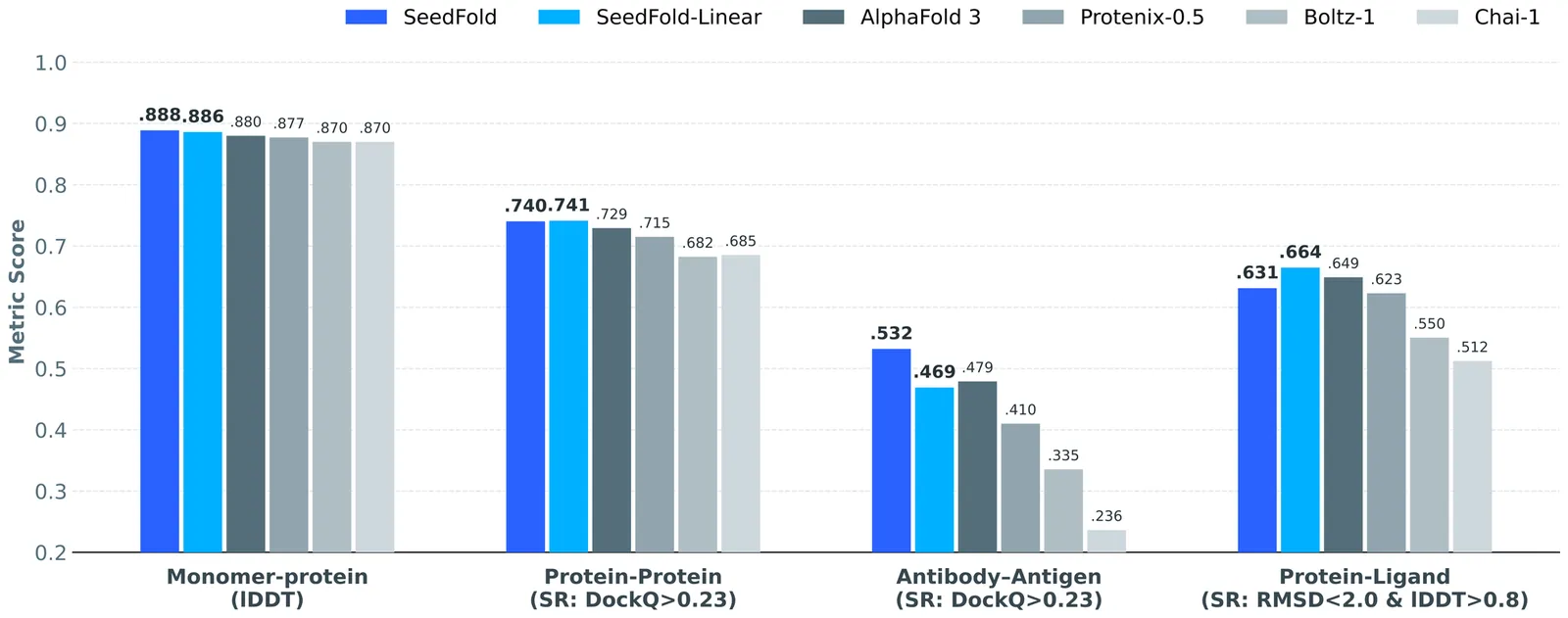
SeedFold: Scaling Biomolecular Structure Prediction
Highly accurate biomolecular structure prediction is a key component of developing biomolecular foundation models, and one of the most critical aspects of building foundation models is identifying the recipes for scaling the model. In this work, we present SeedFold, a folding model that successfully scales up the model capacity. Our contributions are threefold: first, we identify an effective width-scaling strategy for the Pairformer to increase representation capacity; second, we introduce a novel linear triangular attention that reduces computational complexity to enable efficient scaling; finally, we construct a large-scale distillation dataset to substantially enlarge the training set. Experiments on FoldBench show that SeedFold outperforms AlphaFold3 on most protein-related tasks.
2512.243541
Dec 2025Biomolecules
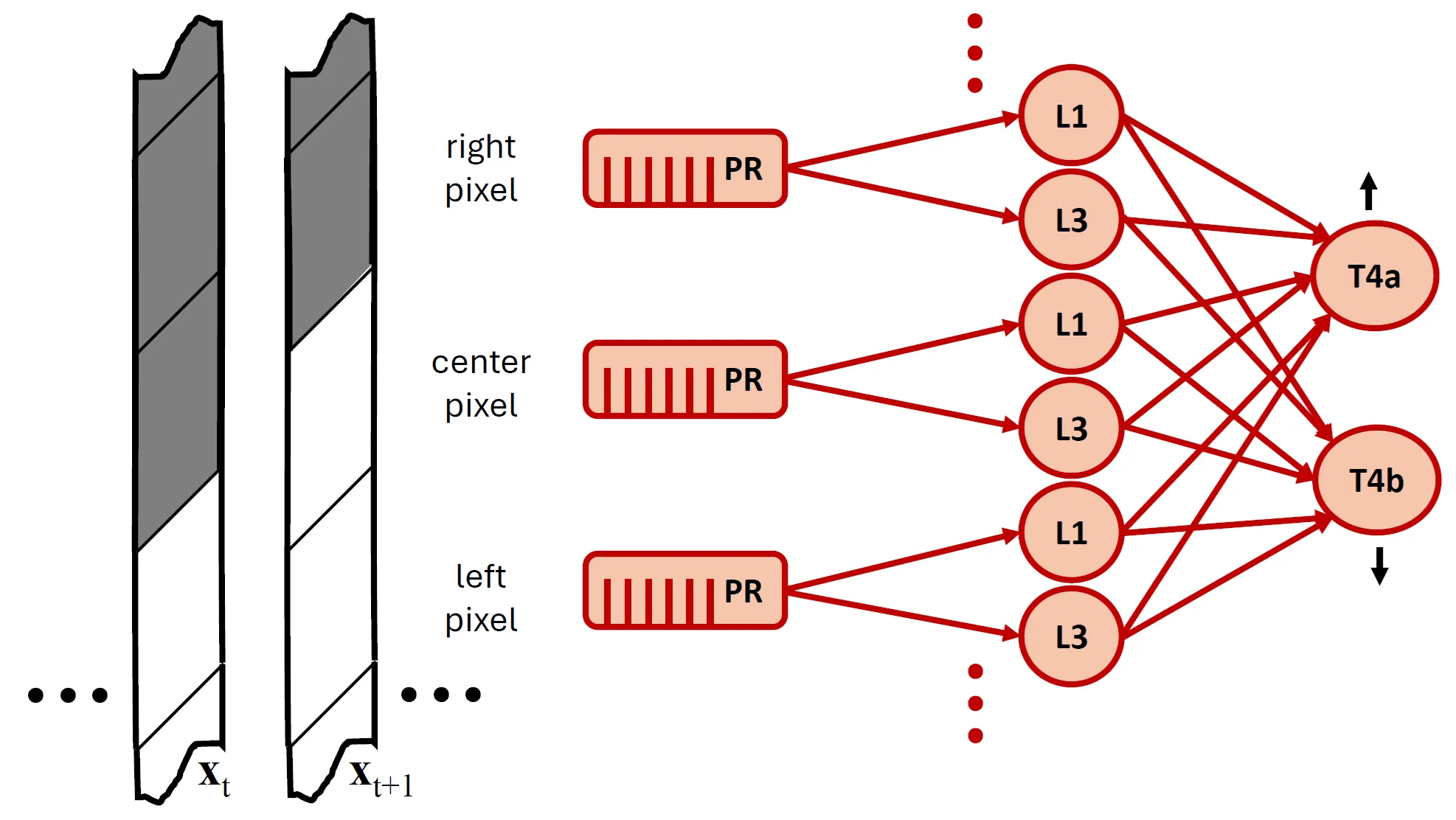
A Network of Biologically Inspired Rectified Spectral Units (ReSUs) Learns Hierarchical Features Without Error Backpropagation
We introduce a biologically inspired, multilayer neural architecture composed of Rectified Spectral Units (ReSUs). Each ReSU projects a recent window of its input history onto a canonical direction obtained via canonical correlation analysis (CCA) of previously observed past-future input pairs, and then rectifies either its positive or negative component. By encoding canonical directions in synaptic weights and temporal filters, ReSUs implement a local, self-supervised algorithm for progressively constructing increasingly complex features. To evaluate both computational power and biological fidelity, we trained a two-layer ReSU network in a self-supervised regime on translating natural scenes. First-layer units, each driven by a single pixel, developed temporal filters resembling those of Drosophila post-photoreceptor neurons (L1/L2 and L3), including their empirically observed adaptation to signal-to-noise ratio (SNR). Second-layer units, which pooled spatially over the first layer, became direction-selective -- analogous to T4 motion-detecting cells -- with learned synaptic weight patterns approximating those derived from connectomic reconstructions. Together, these results suggest that ReSUs offer (i) a principled framework for modeling sensory circuits and (ii) a biologically grounded, backpropagation-free paradigm for constructing deep self-supervised neural networks.
2512.23146
Dec 2025Neurons and Cognition
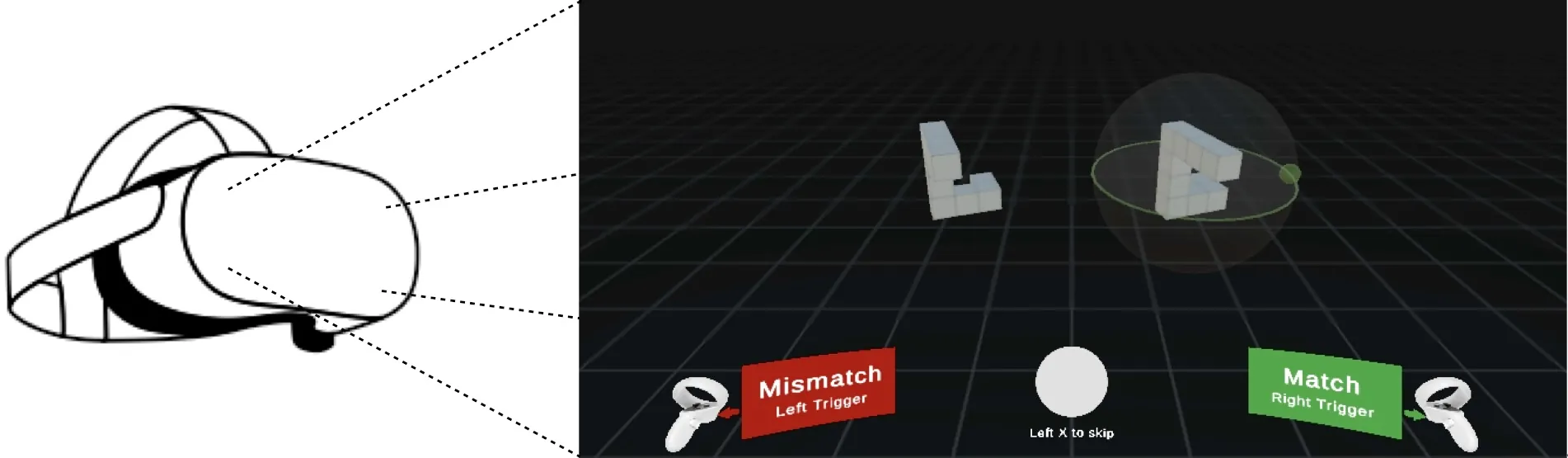
A Deep Learning Model of Mental Rotation Informed by Interactive VR Experiments
Mental rotation -- the ability to compare objects seen from different viewpoints -- is a fundamental example of mental simulation and spatial world modelling in humans. Here we propose a mechanistic model of human mental rotation, leveraging advances in deep, equivariant, and neuro-symbolic learning. Our model consists of three stacked components: (1) an equivariant neural encoder, taking images as input and producing 3D spatial representations of objects, (2) a neuro-symbolic object encoder, deriving symbolic descriptions of objects from these spatial representations, and (3) a neural decision agent, comparing these symbolic descriptions to prescribe rotation simulations in 3D latent space via a recurrent pathway. Our model design is guided by the abundant experimental literature on mental rotation, which we complemented with experiments in VR where participants could at times manipulate the objects to compare, providing us with additional insights into the cognitive process of mental rotation. Our model captures well the performance, response times and behavior of participants in our and others' experiments. The necessity of each model component is shown through systematic ablations. Our work adds to a recent collection of deep neural models of human spatial reasoning, further demonstrating the potency of integrating deep, equivariant, and symbolic representations to model the human mind.
2512.13517
Dec 2025Neurons and Cognition
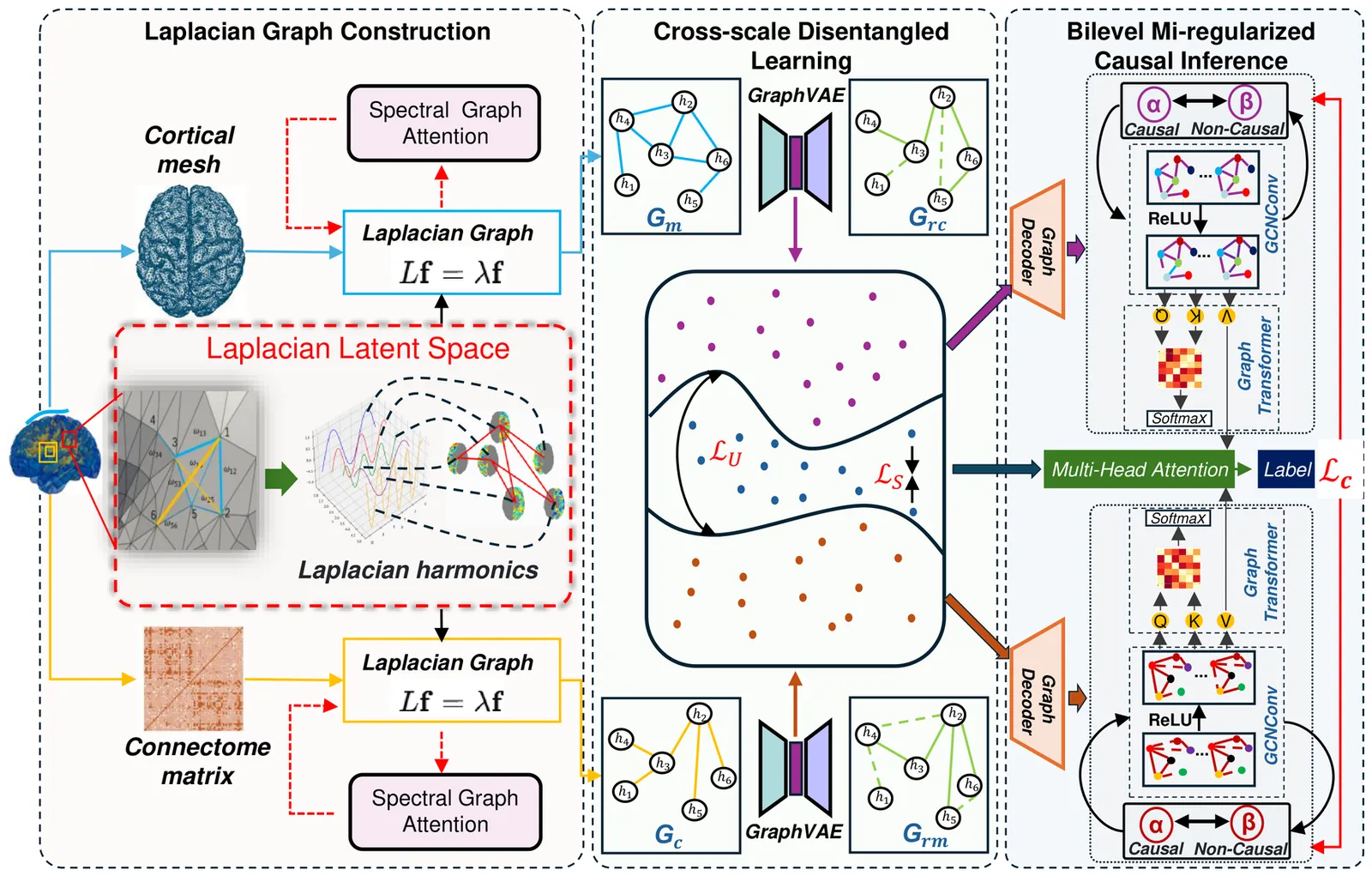
Multiscale Causal Geometric Deep Learning for Modeling Brain Structure
Multimodal MRI offers complementary multi-scale information to characterize the brain structure. However, it remains challenging to effectively integrate multimodal MRI while achieving neuroscience interpretability. Here we propose to use Laplacian harmonics and spectral graph theory for multimodal alignment and multiscale integration. Based on the cortical mesh and connectome matrix that offer multi-scale representations, we devise Laplacian operators and spectral graph attentions to construct a shared latent space for model alignment. Next, we employ a disentangled learning combined with Graph Variational Autoencoder architectures to separate scale-specific and shared features. Lastly, we design a mutual information-informed bilevel regularizer to separate causal and non-causal factors based on the disentangled features, achieving robust model performance with enhanced interpretability. Our model outperforms baselines and other state-of-the-art models. The ablation studies confirmed the effectiveness of the proposed modules. Our model promises to offer a robust and interpretable framework for multi-scale brain structure analysis.
2512.11738
Dec 2025Neurons and Cognition
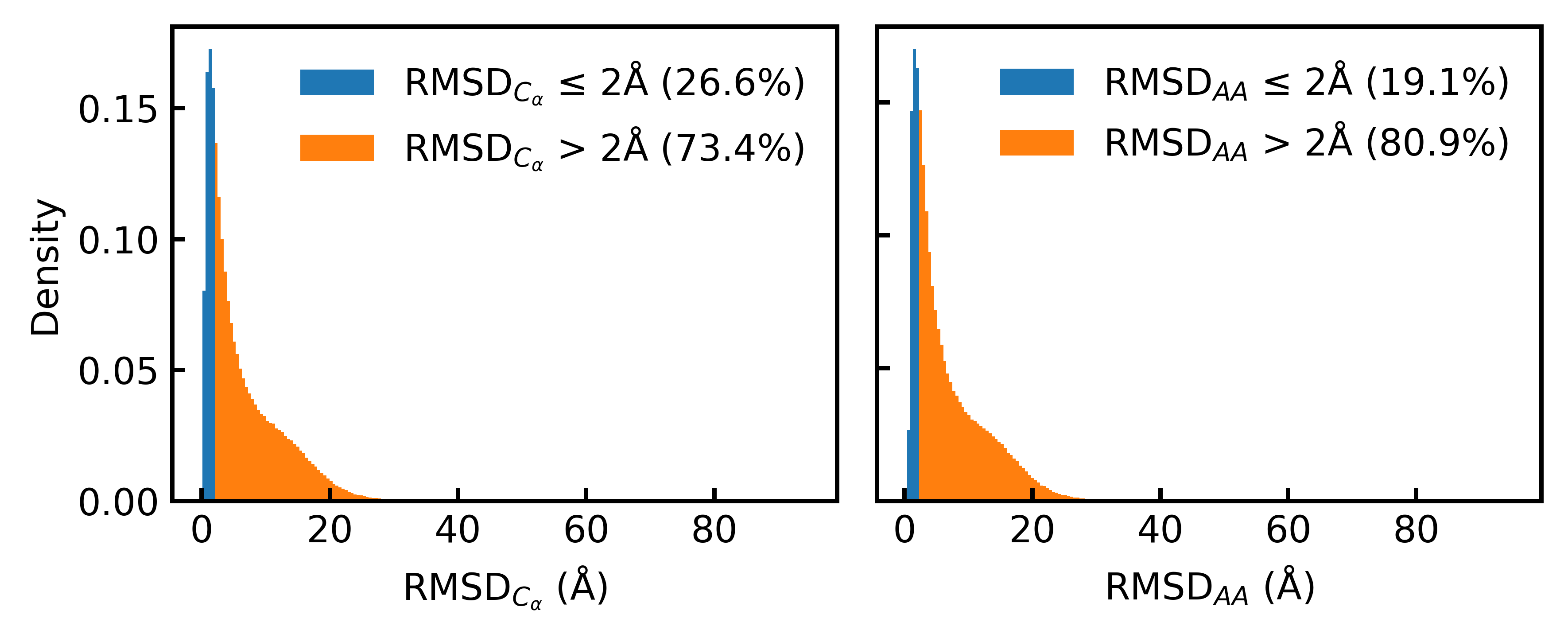
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteina, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteina Atomistica, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving \method's structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.
2512.01976
Dec 2025Biomolecules