Quantitative Methods
arXiv:q-bio.QM
Experimental methods and tools, mathematical methods in biology, bioinformatics.
Looking for a broader view? This category is part of:
Experimental methods and tools, mathematical methods in biology, bioinformatics.
Looking for a broader view? This category is part of:
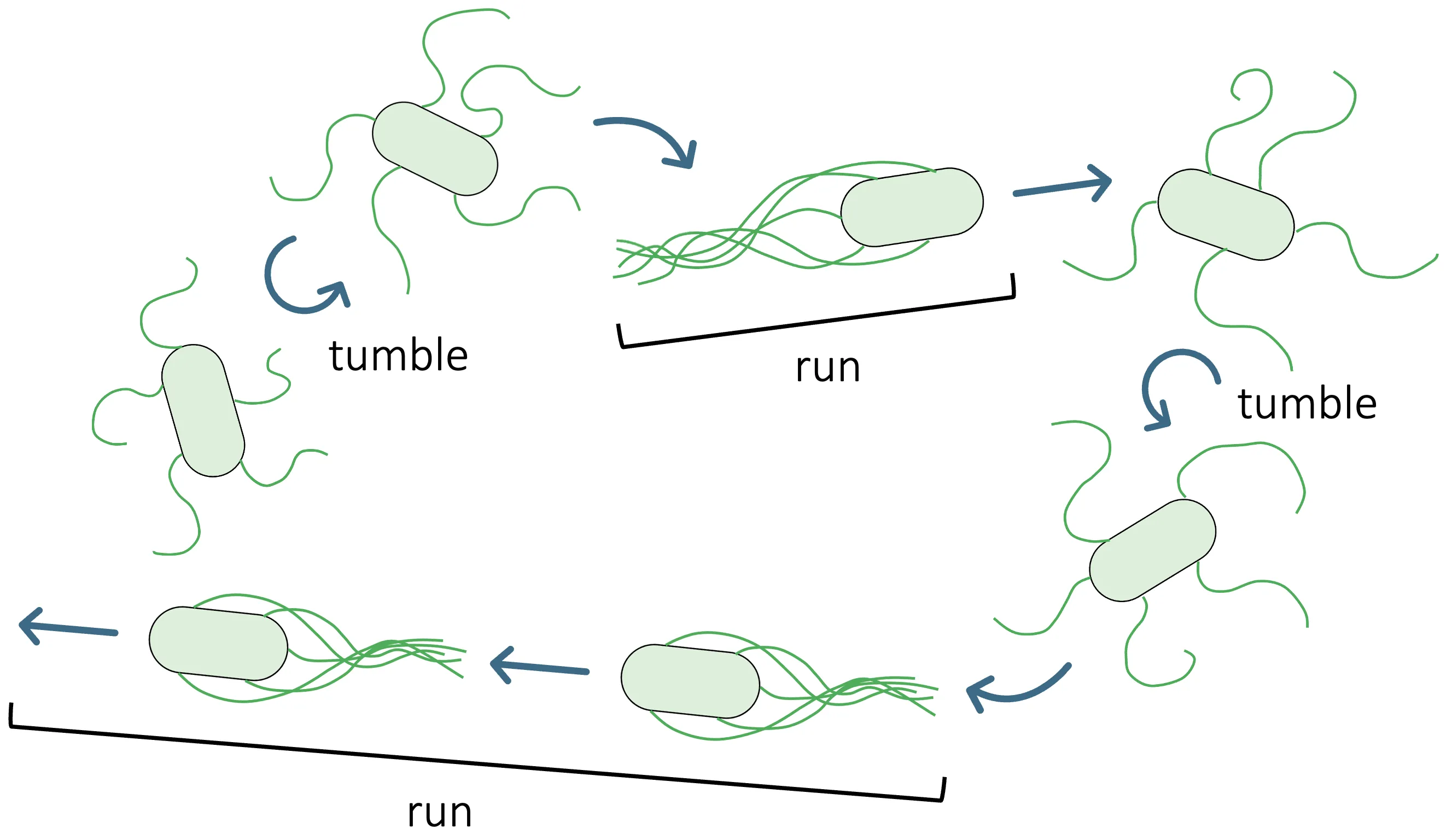
Bacterial chemotactic sensing converts noisy chemical signals into running and tumbling. We analyze the static sensing limits of mixed Tar/Tsr chemoreceptor clusters in individual Escherichia coli cells using a heterogeneous Monod-Wyman-Changeux (MWC) model. By sweeping a seven-dimensional parameter space, we compute three sensing performance metrics-channel capacity, effective Hill coefficient, and dynamic range. Across E. coli-like parameter regimes, we consistently observe pronounced local maxima of channel capacity, whereas neither the effective Hill coefficient nor the dynamic range exhibit comparable optimization. The capacity-achieving input distribution is bimodal, which implies that individual cells maximize information by sampling both low- and high concentration regimes. Together, these results suggest that, at the individual-cell level, channel capacity may be selected for in E. coli receptor clusters.
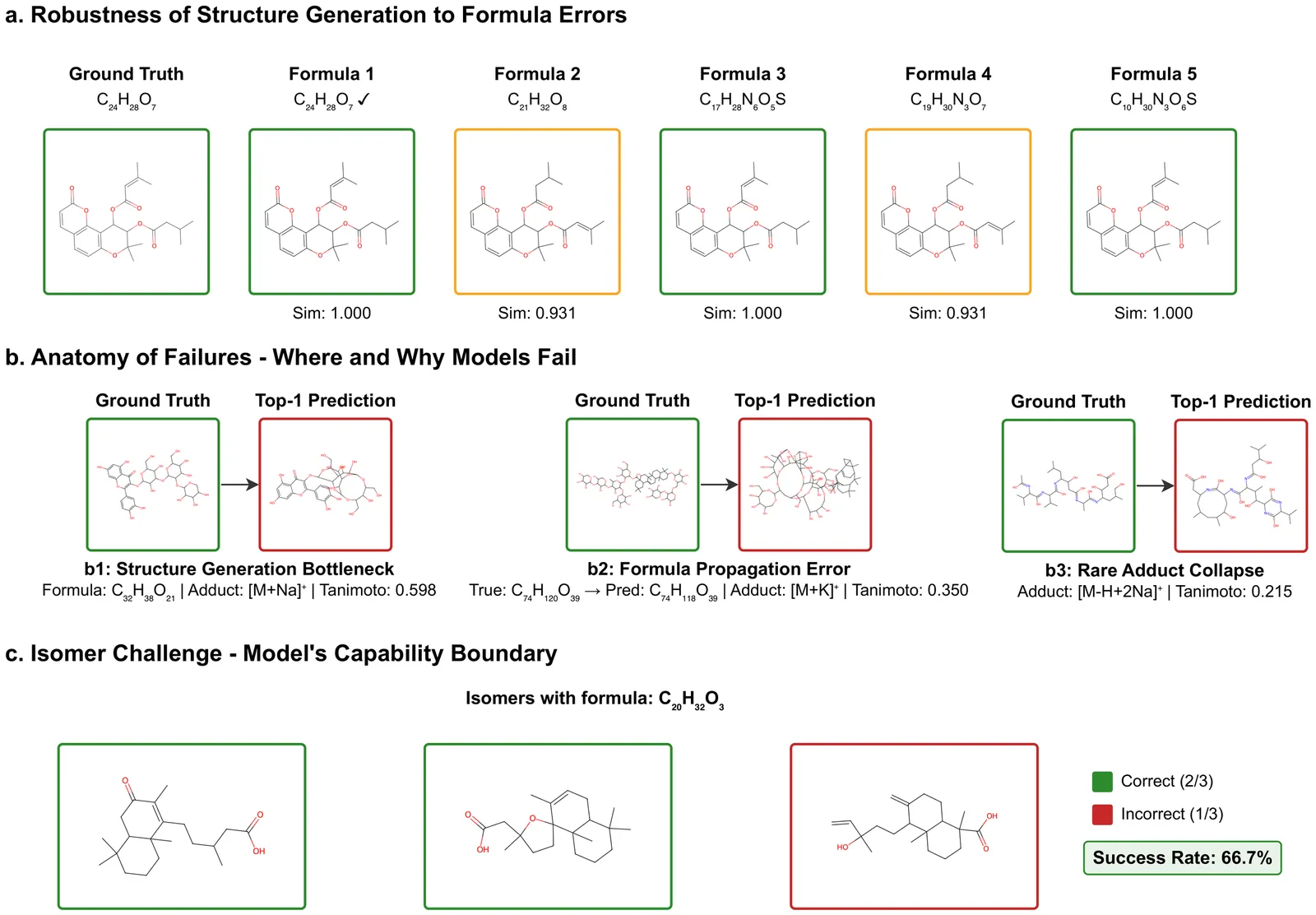
Liquid chromatography mass spectrometry (LC-MS)-based metabolomics and exposomics aim to measure detectable small molecules in biological samples. The results facilitate hypothesis-generating discovery of metabolic changes and disease mechanisms and provide information about environmental exposures and their effects on human health. Metabolomics and exposomics are made possible by the high resolving power of LC and high mass measurement accuracy of MS. However, a majority of the signals from such studies still cannot be identified or annotated using conventional library searching because existing spectral libraries are far from covering the vast chemical space captured by LC-MS/MS. To address this challenge and unleash the full potential of metabolomics and exposomics, a number of computational approaches have been developed to predict compounds based on tandem mass spectra. Published assessment of these approaches used different datasets and evaluation. To select prediction workflows for practical applications and identify areas for further improvements, we have carried out a systematic evaluation of the state-of-the-art prediction algorithms. Specifically, the accuracy of formula prediction and structure prediction was evaluated for different types of adducts. The resulting findings have established realistic performance baselines, identified critical bottlenecks, and provided guidance to further improve compound predictions based on MS.
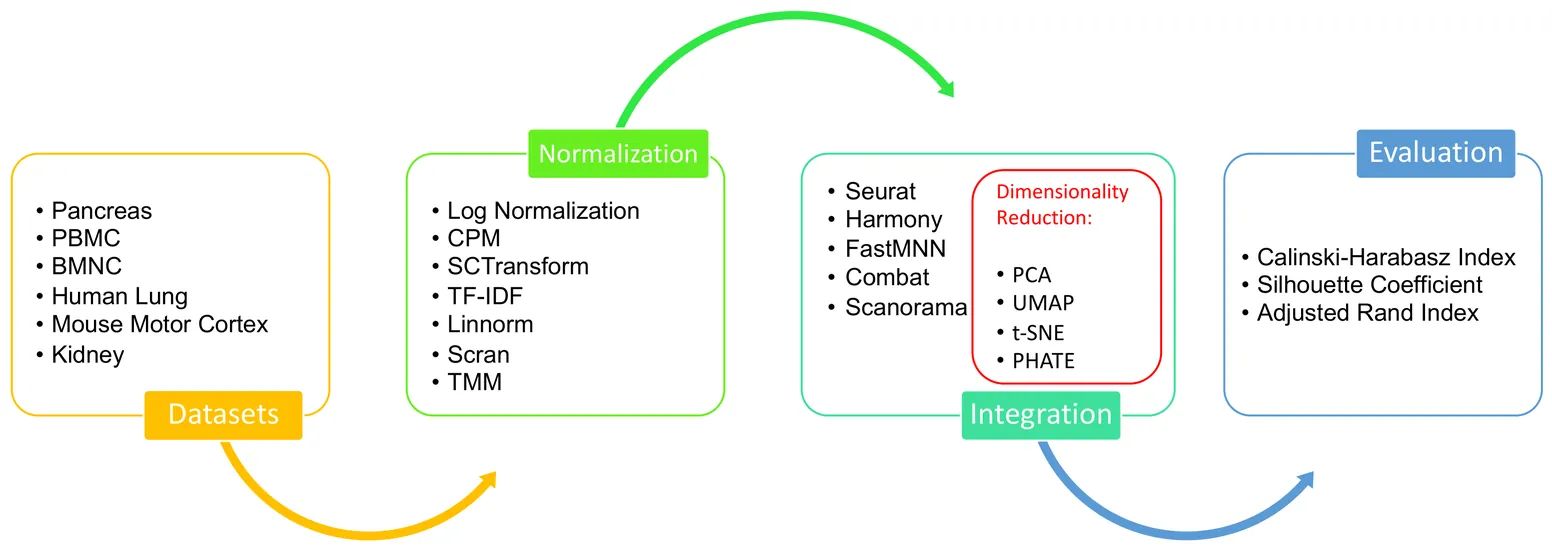
Single-cell data analysis has the potential to revolutionize personalized medicine by characterizing disease-associated molecular changes at the single-cell level. Advanced single-cell multimodal assays can now simultaneously measure various molecules (e.g., DNA, RNA, Protein) across hundreds of thousands of individual cells, providing a comprehensive molecular readout. A significant analytical challenge is integrating single-cell measurements across different modalities. Various methods have been developed to address this challenge, but there has been no systematic evaluation of these techniques with different preprocessing strategies. This study examines a general pipeline for single-cell data analysis, which includes normalization, data integration, and dimensionality reduction. The performance of different algorithm combinations often depends on the dataset sizes and characteristics. We evaluate six datasets across diverse modalities, tissues, and organisms using three metrics: Silhouette Coefficient Score, Adjusted Rand Index, and Calinski-Harabasz Index. Our experiments involve combinations of seven normalization methods, four dimensional reduction methods, and five integration methods. The results show that Seurat and Harmony excel in data integration, with Harmony being more time-efficient, especially for large datasets. UMAP is the most compatible dimensionality reduction method with the integration techniques, and the choice of normalization method varies depending on the integration method used.
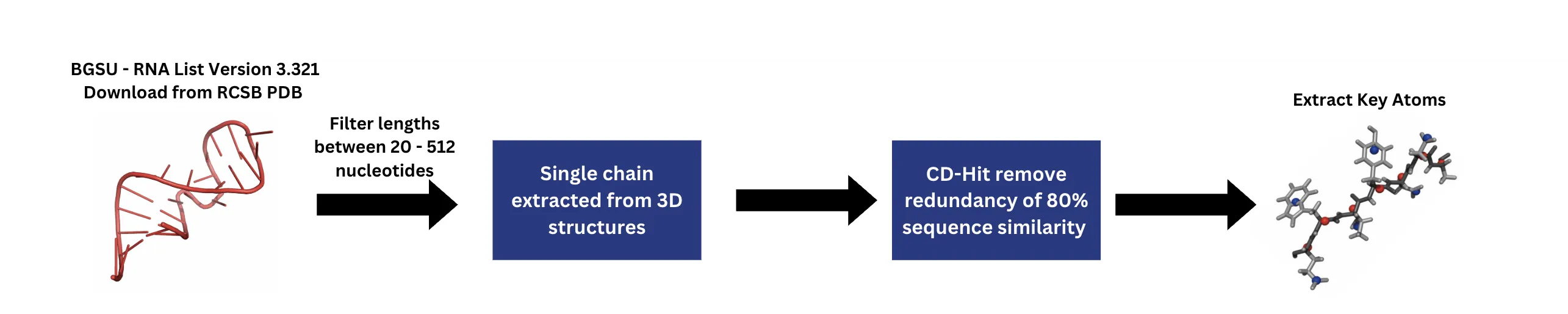
RNA's diverse biological functions stem from its structural versatility, yet accurately predicting and designing RNA sequences given a 3D conformation (inverse folding) remains a challenge. Here, I introduce a deep learning framework that integrates Geometric Vector Perceptron (GVP) layers with a Transformer architecture to enable end-to-end RNA design. I construct a dataset consisting of experimentally solved RNA 3D structures, filtered and deduplicated from the BGSU RNA list, and evaluate performance using both sequence recovery rate and TM-score to assess sequence and structural fidelity, respectively. On standard benchmarks and RNA-Puzzles, my model achieves state-of-the-art performance, with recovery and TM-scores of 0.481 and 0.332, surpassing existing methods across diverse RNA families and length scales. Masked family-level validation using Rfam annotations confirms strong generalization beyond seen families. Furthermore, inverse-folded sequences, when refolded using AlphaFold3, closely resemble native structures, highlighting the critical role of geometric features captured by GVP layers in enhancing Transformer-based RNA design.
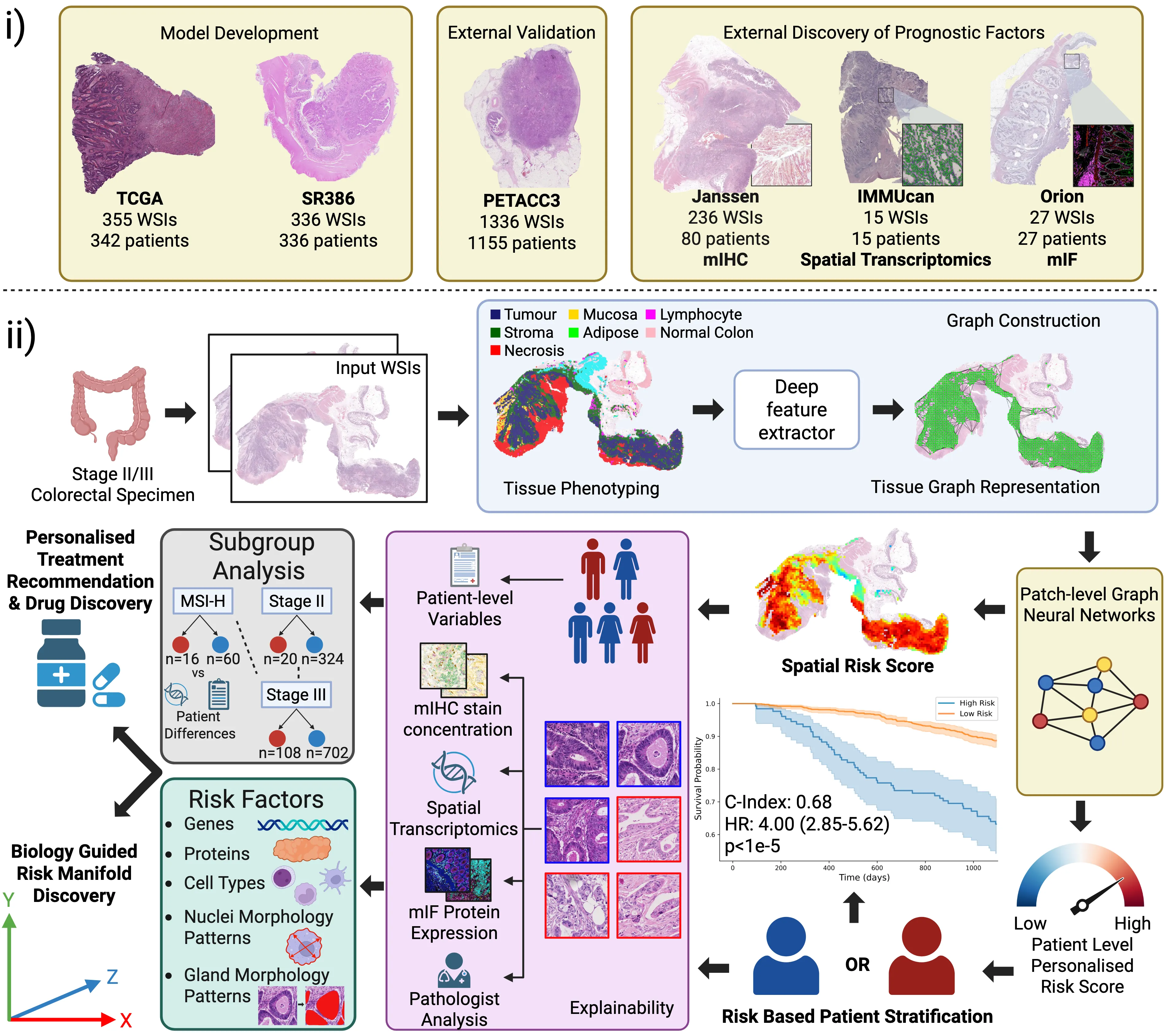
Routine histology contains rich prognostic information in stage II/III colorectal cancer, much of which is embedded in complex spatial tissue organisation. We present INSIGHT, a graph neural network that predicts survival directly from routine histology images. Trained and cross-validated on TCGA (n=342) and SURGEN (n=336), INSIGHT produces patient-level spatially resolved risk scores. Large independent validation showed superior prognostic performance compared with pTNM staging (C-index 0.68-0.69 vs 0.44-0.58). INSIGHT spatial risk maps recapitulated canonical prognostic histopathology and identified nuclear solidity and circularity as quantitative risk correlates. Integrating spatial risk with data-driven spatial transcriptomic signatures, spatial proteomics, bulk RNA-seq, and single-cell references revealed an epithelium-immune risk manifold capturing epithelial dedifferentiation and fetal programs, myeloid-driven stromal states including $\mathrm{SPP1}^{+}$ macrophages and $\mathrm{LAMP3}^{+}$ dendritic cells, and adaptive immune dysfunction. This analysis exposed patient-specific epithelial heterogeneity, stratification within MSI-High tumours, and high-risk routes of CDX2/HNF4A loss and CEACAM5/6-associated proliferative programs, highlighting coordinated therapeutic vulnerabilities.
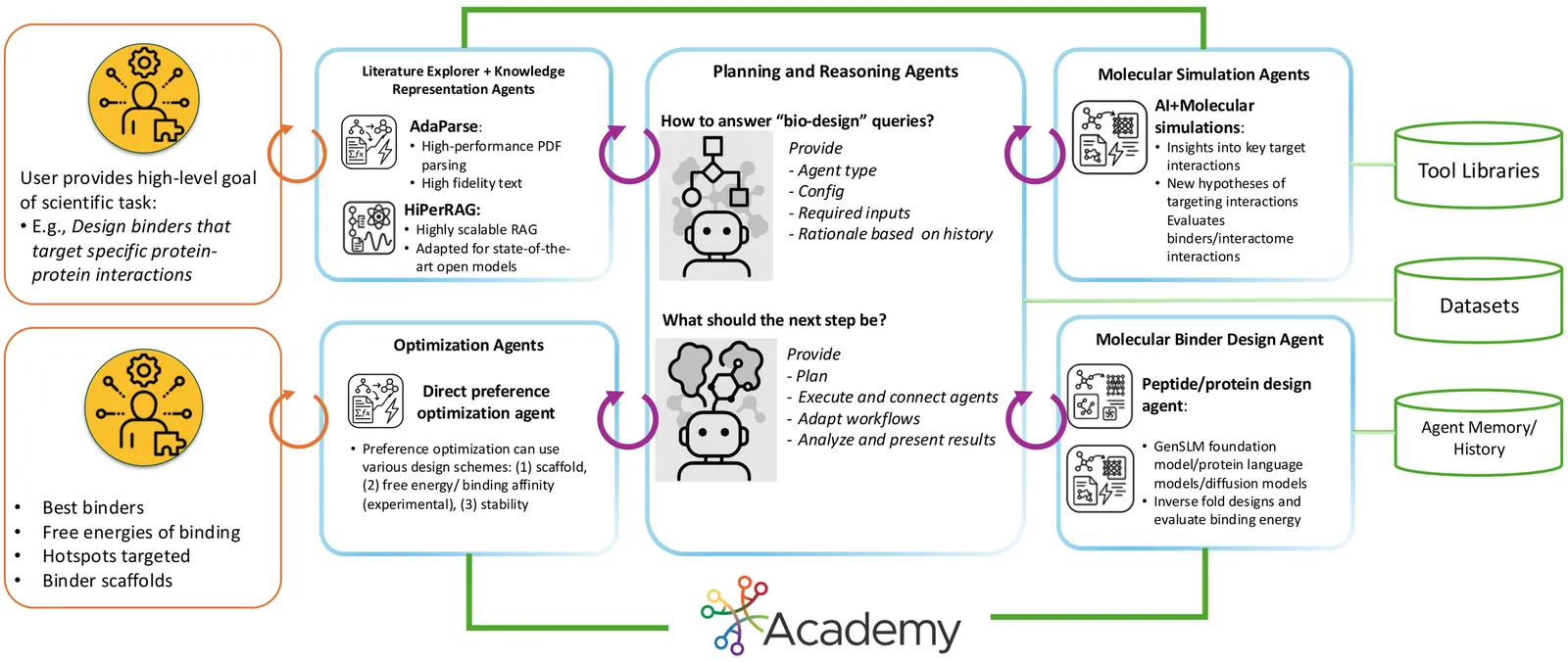
Intrinsically disordered proteins (IDPs) represent crucial therapeutic targets due to their significant role in disease -- approximately 80\% of cancer-related proteins contain long disordered regions -- but their lack of stable secondary/tertiary structures makes them "undruggable". While recent computational advances, such as diffusion models, can design high-affinity IDP binders, translating these to practical drug discovery requires autonomous systems capable of reasoning across complex conformational ensembles and orchestrating diverse computational tools at scale.To address this challenge, we designed and implemented StructBioReasoner, a scalable multi-agent system for designing biologics that can be used to target IDPs. StructBioReasoner employs a novel tournament-based reasoning framework where specialized agents compete to generate and refine therapeutic hypotheses, naturally distributing computational load for efficient exploration of the vast design space. Agents integrate domain knowledge with access to literature synthesis, AI-structure prediction, molecular simulations, and stability analysis, coordinating their execution on HPC infrastructure via an extensible federated agentic middleware, Academy. We benchmark StructBioReasoner across Der f 21 and NMNAT-2 and demonstrate that over 50\% of 787 designed and validated candidates for Der f 21 outperformed the human-designed reference binders from literature, in terms of improved binding free energy. For the more challenging NMNAT-2 protein, we identified three binding modes from 97,066 binders, including the well-studied NMNAT2:p53 interface. Thus, StructBioReasoner lays the groundwork for agentic reasoning systems for IDP therapeutic discovery on Exascale platforms.
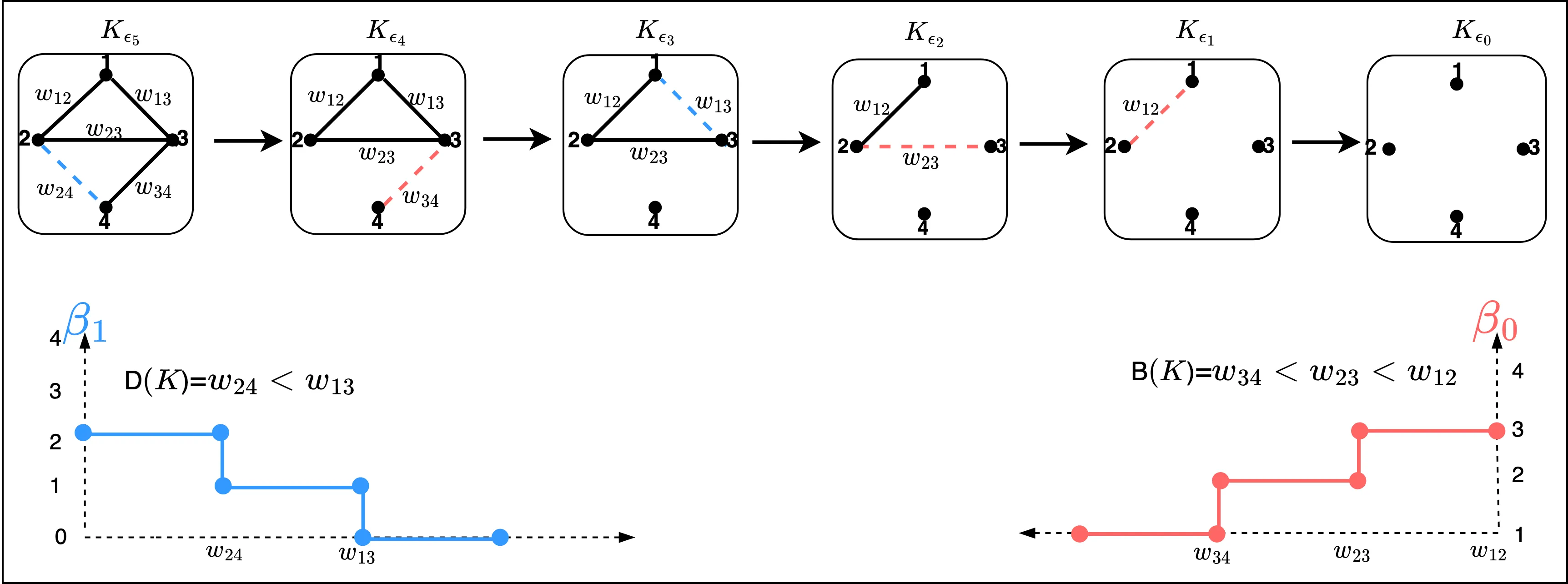
Identifying and comparing topological features, particularly cycles, across different topological objects remains a fundamental challenge in persistent homology and topological data analysis. This work introduces a novel framework for constructing cycle communities through two complementary approaches. First, a dendrogram-based methodology leverages merge-tree algorithms to construct hierarchical representations of homology classes from persistence intervals. The Wasserstein distance on merge trees is introduced as a metric for comparing dendrograms, establishing connections to hierarchical clustering frameworks. Through simulation studies, the discriminative power of dendrogram representations for identifying cycle communities is demonstrated. Second, an extension of Stratified Gradient Sampling simultaneously learns multiple filter functions that yield cycle barycenter functions capable of faithfully reconstructing distinct sets of cycles. The set of cycles each filter function can reconstruct constitutes cycle communities that are non-overlapping and partition the space of all cycles. Together, these approaches transform the problem of cycle matching into both a hierarchical clustering and topological optimization framework, providing principled methods to identify similar topological structures both within and across groups of topological objects.
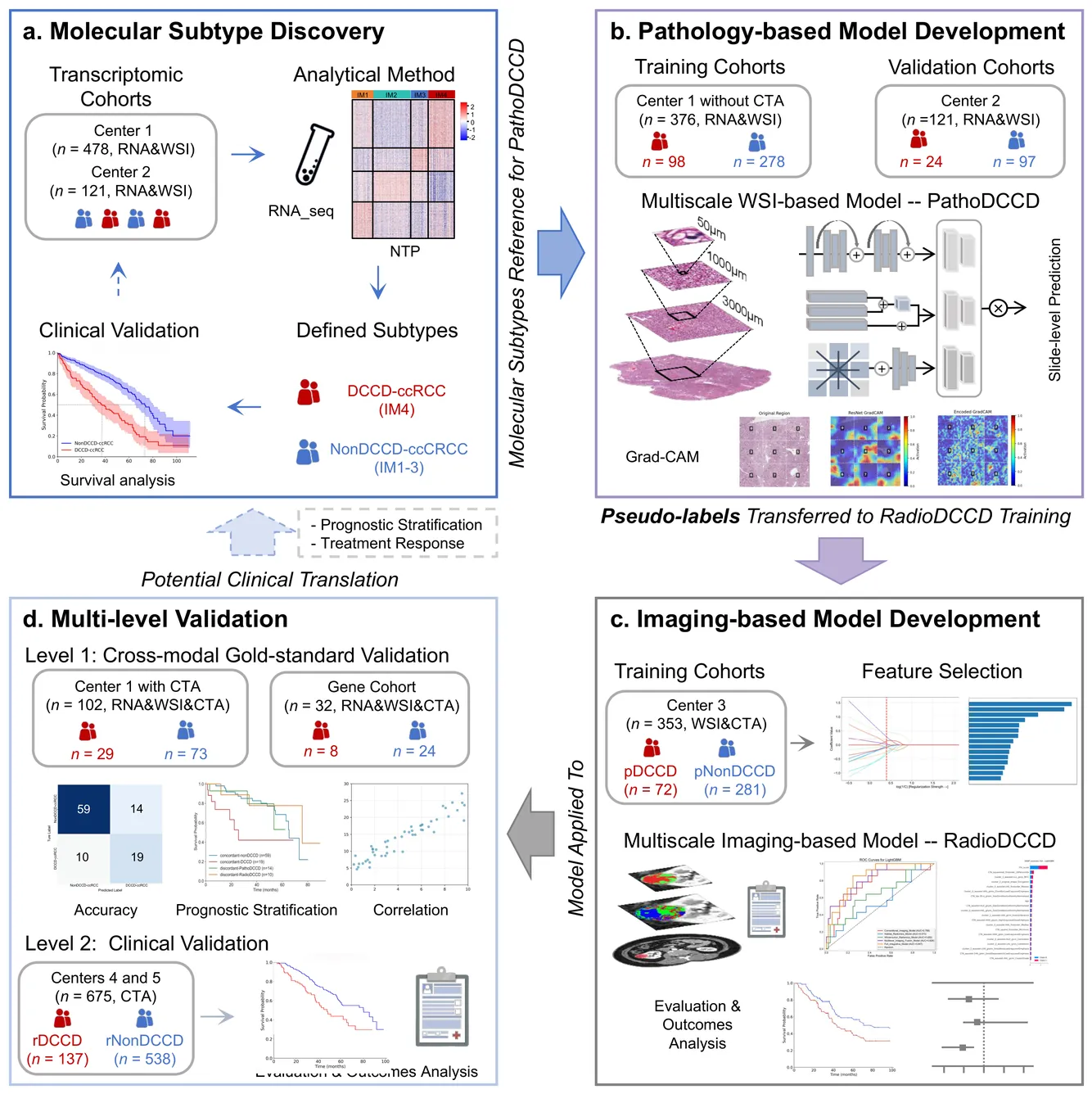
Clear cell renal cell carcinoma (ccRCC) exhibits extensive intratumoral heterogeneity on multiple biological scales, contributing to variable clinical outcomes and limiting the effectiveness of conventional TNM staging, which highlights the urgent need for multiscale integrative analytic frameworks. The lipid-deficient de-clear cell differentiated (DCCD) ccRCC subtype, defined by multi-omics analyses, is associated with adverse outcomes even in early-stage disease. Here, we establish a hierarchical cross-scale framework for the preoperative identification of DCCD-ccRCC. At the highest layer, cross-modal mapping transferred molecular signatures to histological and CT phenotypes, establishing a molecular-to-pathology-to-radiology supervisory bridge. Within this framework, each modality-specific model is designed to mirror the inherent hierarchical structure of tumor biology. PathoDCCD captured multi-scale microscopic features, from cellular morphology and tissue architecture to meso-regional organization. RadioDCCD integrated complementary macroscopic information by combining whole-tumor and its habitat-subregions radiomics with a 2D maximal-section heterogeneity metric. These nested models enabled integrated molecular subtype prediction and clinical risk stratification. Across five cohorts totaling 1,659 patients, PathoDCCD reliably recapitulated molecular subtypes, while RadioDCCD provided reliable preoperative prediction. The consistent predictions identified patients with the poorest clinical outcomes. This cross-scale paradigm unifies molecular biology, computational pathology, and quantitative radiology into a biologically grounded strategy for preoperative noninvasive molecular phenotyping of ccRCC.
Post-translational modifications (PTMs) serve as a dynamic chemical language regulating protein function, yet current proteomic methods remain blind to a vast portion of the modified proteome. Standard database search algorithms suffer from a combinatorial explosion of search spaces, limiting the identification of uncharacterized or complex modifications. Here we introduce OmniNovo, a unified deep learning framework for reference-free sequencing of unmodified and modified peptides directly from tandem mass spectra. Unlike existing tools restricted to specific modification types, OmniNovo learns universal fragmentation rules to decipher diverse PTMs within a single coherent model. By integrating a mass-constrained decoding algorithm with rigorous false discovery rate estimation, OmniNovo achieves state-of-the-art accuracy, identifying 51\% more peptides than standard approaches at a 1\% false discovery rate. Crucially, the model generalizes to biological sites unseen during training, illuminating the dark matter of the proteome and enabling unbiased comprehensive analysis of cellular regulation.
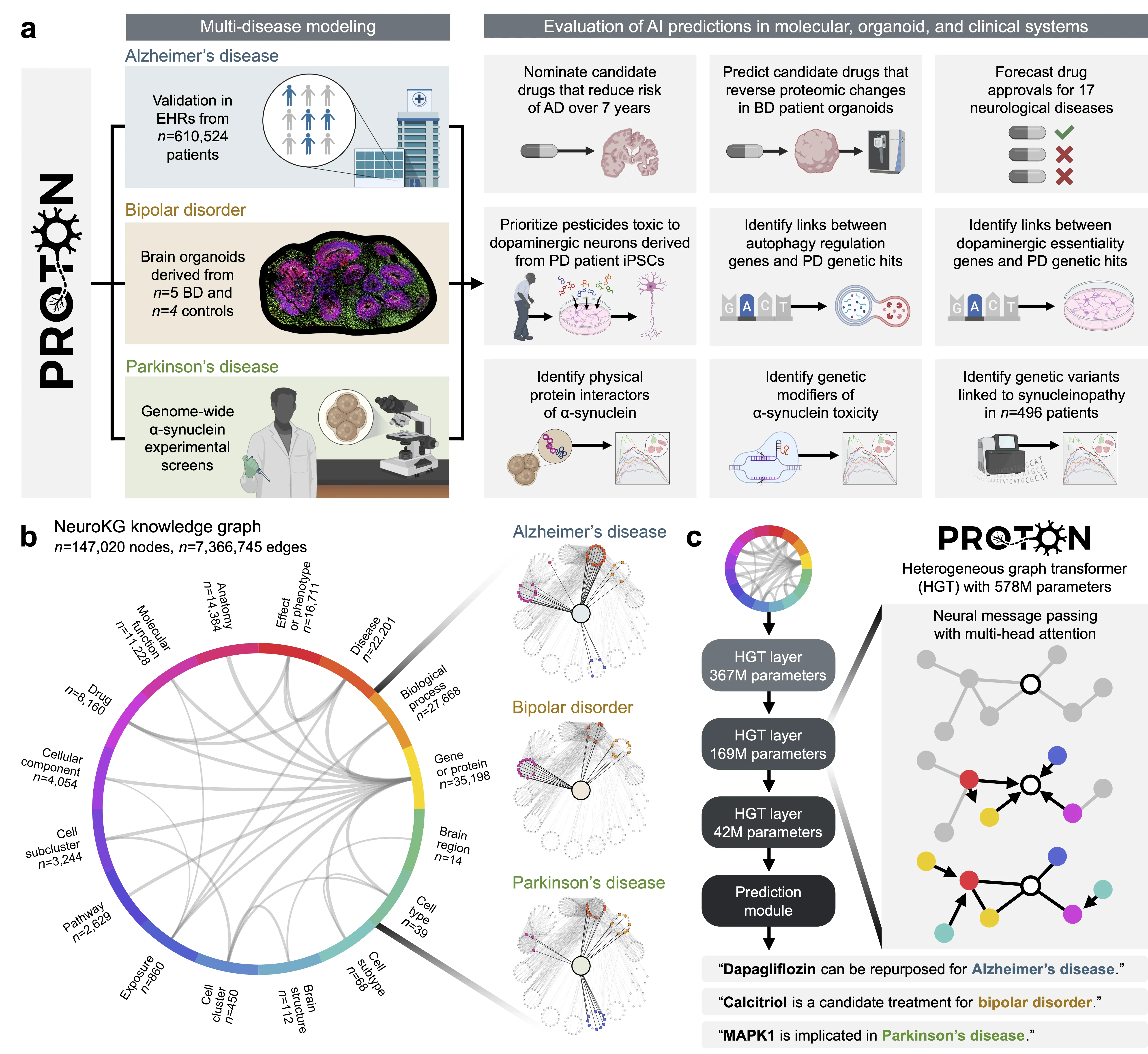
Neurological diseases are the leading global cause of disability, yet most lack disease-modifying treatments. We present PROTON, a heterogeneous graph transformer that generates testable hypotheses across molecular, organoid, and clinical systems. To evaluate PROTON, we apply it to Parkinson's disease (PD), bipolar disorder (BD), and Alzheimer's disease (AD). In PD, PROTON linked genetic risk loci to genes essential for dopaminergic neuron survival and predicted pesticides toxic to patient-derived neurons, including the insecticide endosulfan, which ranked within the top 1.29% of predictions. In silico screens performed by PROTON reproduced six genome-wide $α$-synuclein experiments, including a split-ubiquitin yeast two-hybrid system (normalized enrichment score [NES] = 2.30, FDR-adjusted $p < 1 \times 10^{-4}$), an ascorbate peroxidase proximity labeling assay (NES = 2.16, FDR $< 1 \times 10^{-4}$), and a high-depth targeted exome sequencing study in 496 synucleinopathy patients (NES = 2.13, FDR $< 1 \times 10^{-4}$). In BD, PROTON predicted calcitriol as a candidate drug that reversed proteomic alterations observed in cortical organoids derived from BD patients. In AD, we evaluated PROTON predictions in health records from $n = 610,524$ patients at Mass General Brigham, confirming that five PROTON-predicted drugs were associated with reduced seven-year dementia risk (minimum hazard ratio = 0.63, 95% CI: 0.53-0.75, $p < 1 \times 10^{-7}$). PROTON generated neurological hypotheses that were evaluated across molecular, organoid, and clinical systems, defining a path for AI-driven discovery in neurological disease.
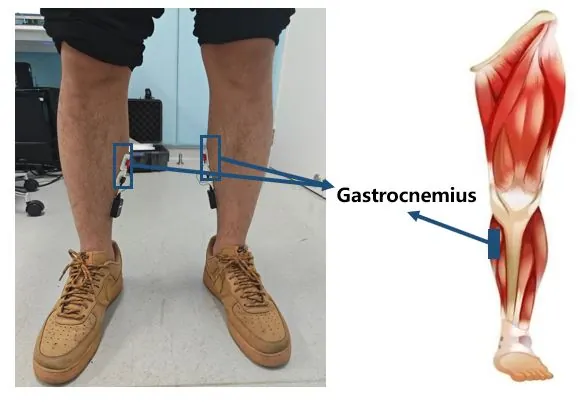
Rehabilitation exoskeletons have shown promising results in promoting recovery for stroke patients. Accurately and timely identifying the motion intentions of patients is a critical challenge in enhancing active participation during lower limb exoskeleton-assisted rehabilitation training. This paper proposes a Dual-Channel Attentive Fusion Network (DCAF-Net) that synergistically integrates pre-movement surface electromyography (sEMG) and inertial measurement unit (IMU) data for lower limb intention prediction in stroke patients. First, a dual-channel adaptive channel attention module is designed to extract discriminative features from 48 time-domain and frequency-domain features derived from bilateral gastrocnemius sEMG signals. Second, an IMU encoder combining convolutional neural network (CNN) and attention-based long short-term memory (attention-LSTM) layers is designed to decode temporal-spatial movement patterns. Third, the sEMG and IMU features are fused through concatenation to enable accurate recognition of motion intention. Extensive experiment on 11 participants (8 stroke subjects and 3 healthy subjects) demonstrate the effectiveness of DCAF-Net. It achieved a prediction accuracies of 97.19% for patients and 93.56% for healthy subjects. This study provides a viable solution for implementing intention-driven human-in-the-loop assistance control in clinical rehabilitation robotics.
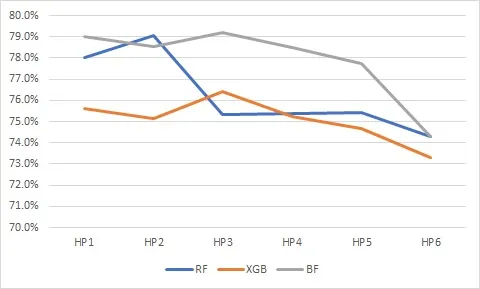
Cancer patients may undergo lengthy and painful chemotherapy treatments, comprising several successive regimens or plans. Treatment inefficacy and other adverse events can lead to discontinuation (or failure) of these plans, or prematurely changing them, which results in a significant amount of physical, financial, and emotional toxicity to the patients and their families. In this work, we build treatment failure models based on the Real World Evidence (RWE) gathered from patients' profiles available in our oncology EMR/EHR system. We also describe our feature engineering pipeline, experimental methods, and valuable insights obtained about treatment failures from trained models. We report our findings on five primary cancer types with the most frequent treatment failures (or discontinuations) to build unique and novel feature vectors from the clinical notes, diagnoses, and medications that are available in our oncology EMR. After following a novel three axes - performance, complexity and explainability - design exploration framework, boosted random forests are selected because they provide a baseline accuracy of 80% and an F1 score of 75%, with reduced model complexity, thus making them more interpretable to and usable by oncologists.
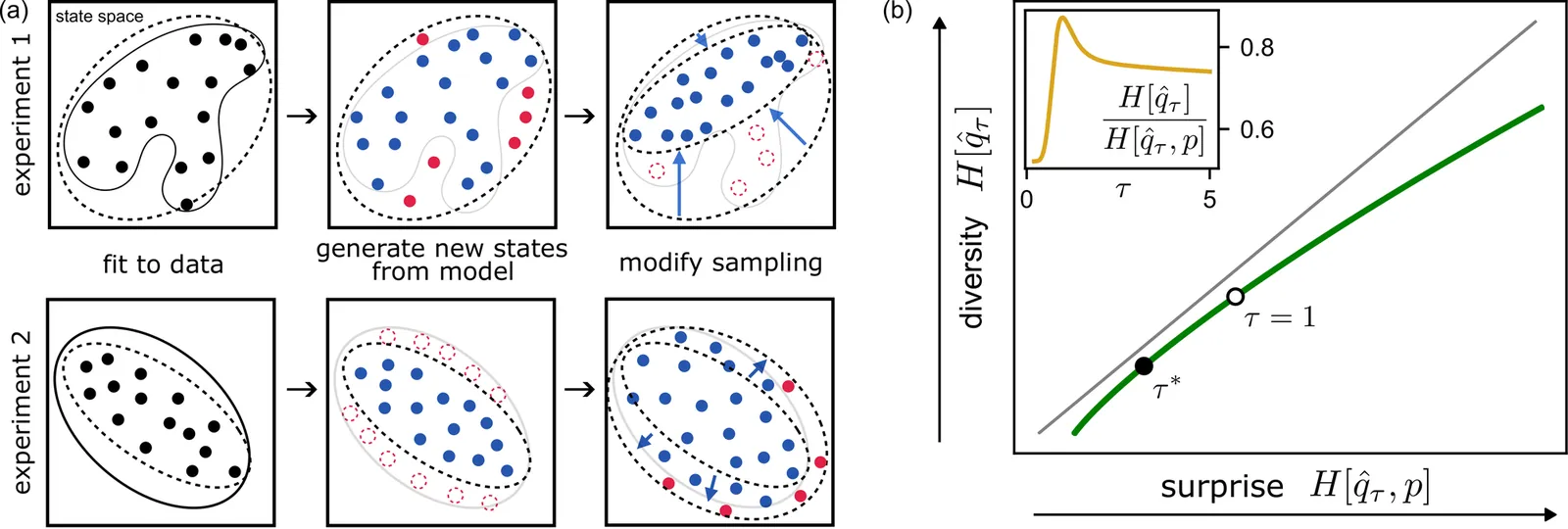
Generative models of complex systems often require post-hoc parameter adjustments to produce useful outputs. For example, energy-based models for protein design are sampled at an artificially low ''temperature'' to generate novel, functional sequences. This temperature tuning is a common yet poorly understood heuristic used across machine learning contexts to control the trade-off between generative fidelity and diversity. Here, we develop an interpretable, physically motivated framework to explain this phenomenon. We demonstrate that in systems with a large ''energy gap'' - separating a small fraction of meaningful states from a vast space of unrealistic states - learning from sparse data causes models to systematically overestimate high-energy state probabilities, a bias that lowering the sampling temperature corrects. More generally, we characterize how the optimal sampling temperature depends on the interplay between data size and the system's underlying energy landscape. Crucially, our results show that lowering the sampling temperature is not always desirable; we identify the conditions where \emph{raising} it results in better generative performance. Our framework thus casts post-hoc temperature tuning as a diagnostic tool that reveals properties of the true data distribution and the limits of the learned model.
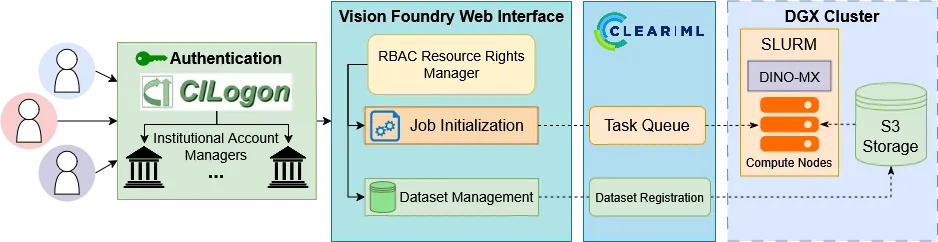
Self-supervised learning (SSL) leverages vast unannotated medical datasets, yet steep technical barriers limit adoption by clinical researchers. We introduce Vision Foundry, a code-free, HIPAA-compliant platform that democratizes pre-training, adaptation, and deployment of foundational vision models. The system integrates the DINO-MX framework, abstracting distributed infrastructure complexities while implementing specialized strategies like Magnification-Aware Distillation (MAD) and Parameter-Efficient Fine-Tuning (PEFT). We validate the platform across domains, including neuropathology segmentation, lung cellularity estimation, and coronary calcium scoring. Our experiments demonstrate that models trained via Vision Foundry significantly outperform generic baselines in segmentation fidelity and regression accuracy, while exhibiting robust zero-shot generalization across imaging protocols. By bridging the gap between advanced representation learning and practical application, Vision Foundry enables domain experts to develop state-of-the-art clinical AI tools with minimal annotation overhead, shifting focus from engineering optimization to clinical discovery.

In multicellular organisms, cells coordinate their activities through cell-cell communication (CCC), which are crucial for development, tissue homeostasis, and disease progression. Recent advances in single-cell and spatial omics technologies provide unprecedented opportunities to systematically infer and analyze CCC from these omics data, either by integrating prior knowledge of ligand-receptor interactions (LRIs) or through de novo approaches. A variety of computational methods have been developed, focusing on methodological innovations, accurate modeling of complex signaling mechanisms, and investigation of broader biological questions. These advances have greatly enhanced our ability to analyze CCC and generate biological hypotheses. Here, we introduce the biological mechanisms and modeling strategies of CCC, and provide a focused overview of more than 140 computational methods for inferring CCC from single-cell and spatial transcriptomic data, emphasizing the diversity in methodological frameworks and biological questions. Finally, we discuss the current challenges and future opportunities in this rapidly evolving field.

In cell culture bioprocessing, real-time batch process monitoring (BPM) refers to the continuous tracking and analysis of key process variables such as viable cell density, nutrient levels, metabolite concentrations, and product titer throughout the duration of a batch run. This enables early detection of deviations and supports timely control actions to ensure optimal cell growth and product quality. BPM plays a critical role in ensuring the quality and regulatory compliance of biopharmaceutical manufacturing processes. However, the development of accurate soft sensors for BPM is hindered by key challenges, including limited historical data, infrequent feedback, heterogeneous process conditions, and high-dimensional sensory inputs. This study presents a comprehensive benchmarking analysis of machine learning (ML) methods designed to address these challenges, with a focus on learning from historical data with limited volume and relevance in the context of bioprocess monitoring. We evaluate multiple ML approaches including feature dimensionality reduction, online learning, and just-in-time learning across three datasets, one in silico dataset and two real-world experimental datasets. Our findings highlight the importance of training strategies in handling limited data and feedback, with batch learning proving effective in homogeneous settings, while just-in-time learning and online learning demonstrate superior adaptability in cold-start scenarios. Additionally, we identify key meta-features, such as feed media composition and process control strategies, that significantly impact model transferability. The results also suggest that integrating Raman-based predictions with lagged offline measurements enhances monitoring accuracy, offering a promising direction for future bioprocess soft sensor development.
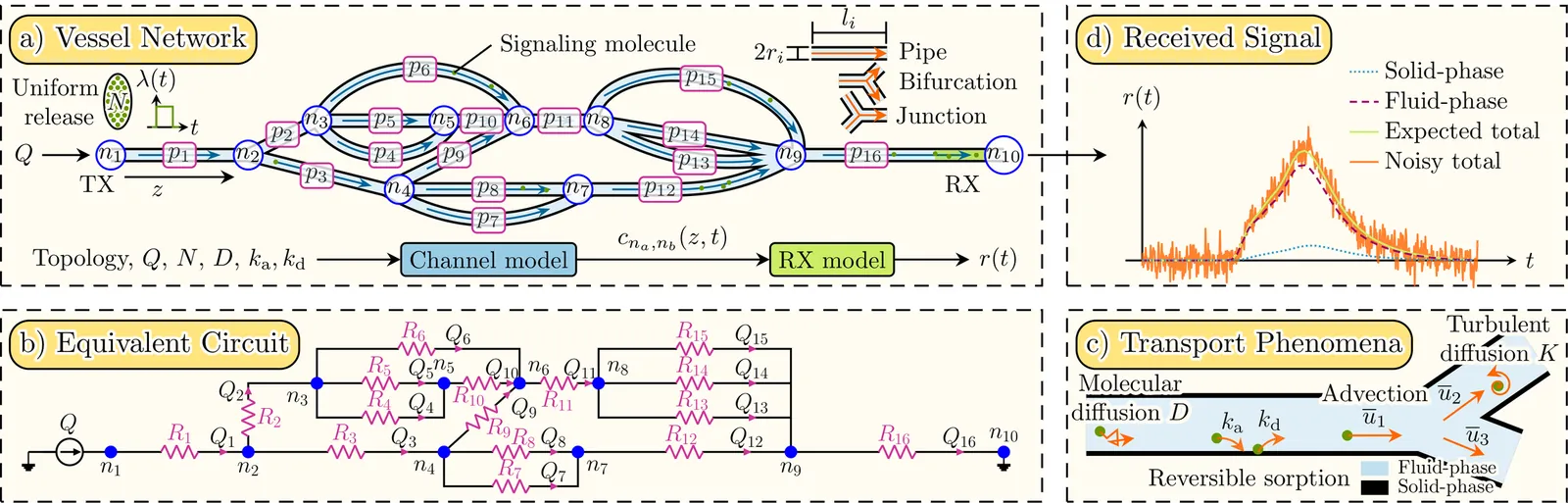
The notion of synthetic molecular communication (MC) refers to the transmission of information via signaling molecules and is foreseen to enable innovative medical applications in the human cardiovascular system (CVS). Crucially, the design of such applications requires accurate and experimentally validated channel models that characterize the propagation of signaling molecules, not just in individual blood vessels, but in complex vessel networks (VNs), as prevalent in the CVS. However, experimentally validated models for MC in VNs remain scarce. To address this gap, we propose a novel channel model for MC in complex VN topologies, which captures molecular transport via advection, molecular and turbulent diffusion, as well as adsorption and desorption at the vessel walls. We specialize this model for superparamagnetic iron-oxide nanoparticles (SPIONs) as signaling molecules by introducing a new receiver (RX) model for planar coil inductive sensors, enabling end-to-end experimental validation with a dedicated SPION testbed. Validation covers a range of channel topologies, from single-vessel topologies to branched VNs with multiple paths between transmitter (TX) and RX. Additionally, to quantify how the VN topology impacts signal quality, and inspired by multi-path propagation models in conventional wireless communications, we introduce two metrics, namely molecule delay and multi-path spread. We show that these metrics link the VN structure to molecule dispersion induced by the VN and mediately to the resulting signal-to-noise ratio (SNR) at the RX. The proposed VN structure-SNR link is validated experimentally, demonstrating that the proposed framework can support tasks such as optimal sensor placement in the CVS or the identification of suitable testbed topologies for specific SNR requirements. All experimental data are openly available on Zenodo.
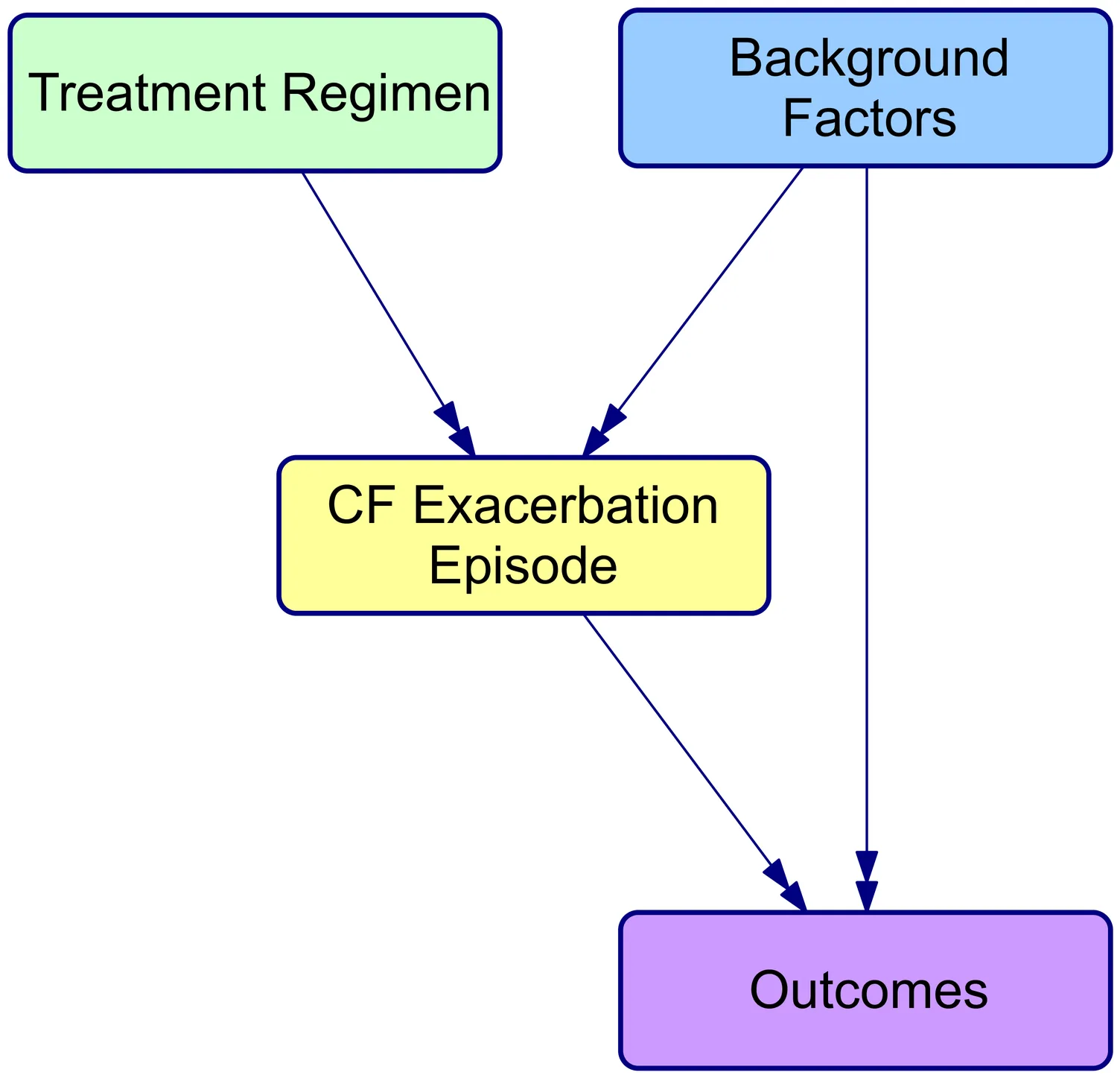
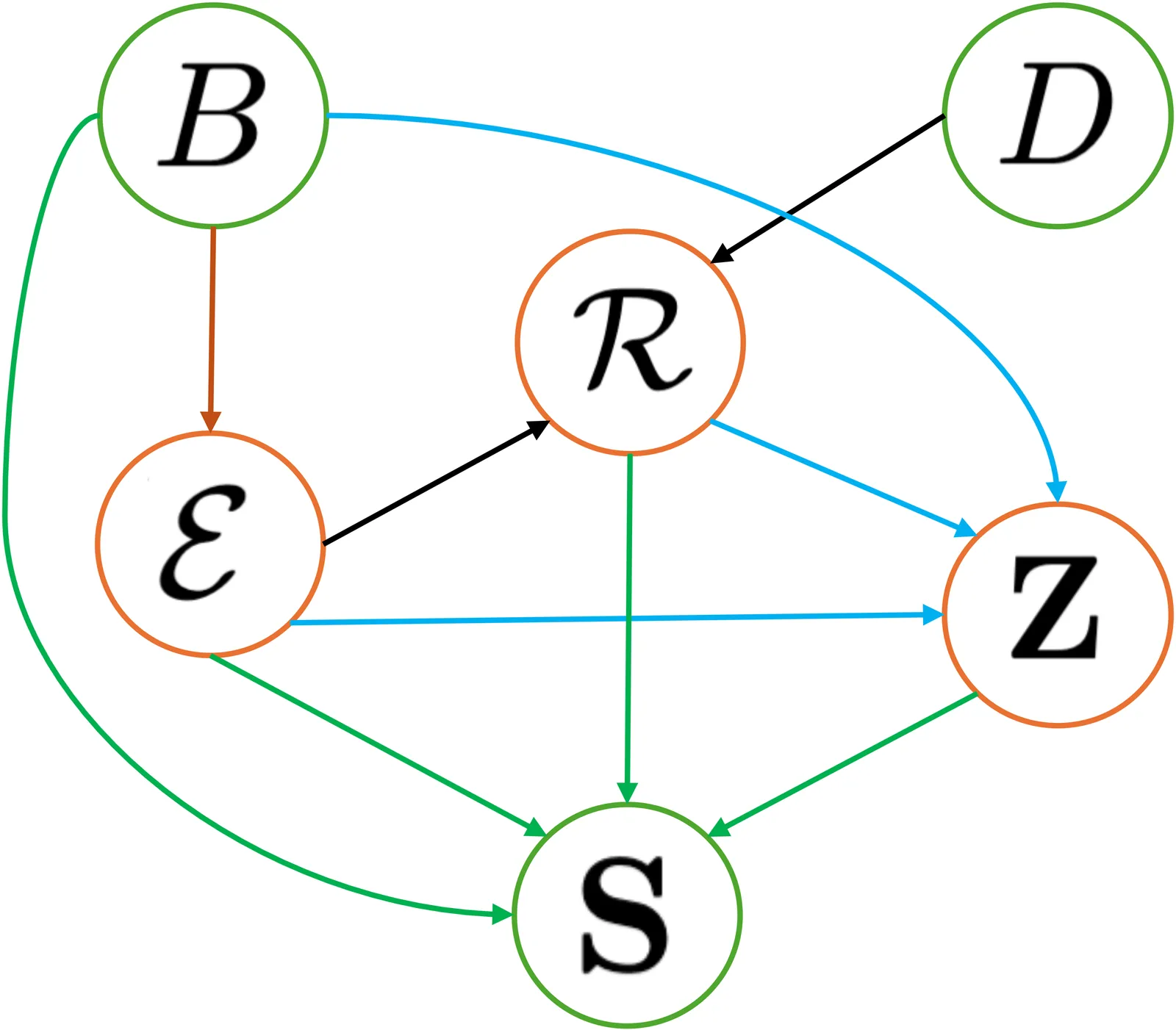
Loss of lung function in cystic fibrosis (CF) occurs progressively, punctuated by acute pulmonary exacerbations (PEx) in which abrupt declines in lung function are not fully recovered. A key component of CF management over the past half century has been the treatment of PEx to slow lung function decline. This has been credited with improvements in survival for people with CF (PwCF), but there is no consensus on the optimal approach to PEx management. BEAT-CF (Bayesian evidence-adaptive treatment of CF) was established to build an evidence-informed knowledge base for CF management. The BEAT-CF causal model is a directed acyclic graph (DAG) and Bayesian network (BN) for PEx that aims to inform the design and analysis of clinical trials comparing the effectiveness of alternative approaches to PEx management. The causal model describes relationships between background risk factors, treatments, and pathogen colonisation of the airways that affect the outcome of an individual PEx episode. The key factors, outcomes, and causal relationships were elicited from CF clinical experts and together represent current expert understanding of the pathophysiology of a PEx episode, guiding the design of data collection and studies and enabling causal inference. Here, we present the DAG that documents this understanding, along with the processes used in its development, providing transparency around our trial design and study processes, as well as a reusable framework for others.

Selecting an effective docking algorithm is highly context-dependent, and no single method performs reliably across structural, chemical, or protocol regimes. We introduce MolAS, a lightweight algorithm selection system that predicts per-algorithm performance from pretrained protein-ligand embeddings using attentional pooling and a shallow residual decoder. With only hundreds to a few thousand labelled complexes, MolAS achieves up to 15% absolute improvement over the single-best solver (SBS) and closes 17-66% of the Virtual Best Solver (VBS)-SBS gap across five diverse docking benchmarks. Analyses of reliability, embedding geometry, and solver-selection patterns show that MolAS succeeds when the oracle landscape exhibits low entropy and separable solver behaviour, but collapses under protocol-induced hierarchy shifts. These findings indicate that the main barrier to robust docking AS is not representational capacity but instability in solver rankings across pose-generation regimes, positioning MolAS as both a practical in-domain selector and a diagnostic tool for assessing when AS is feasible.
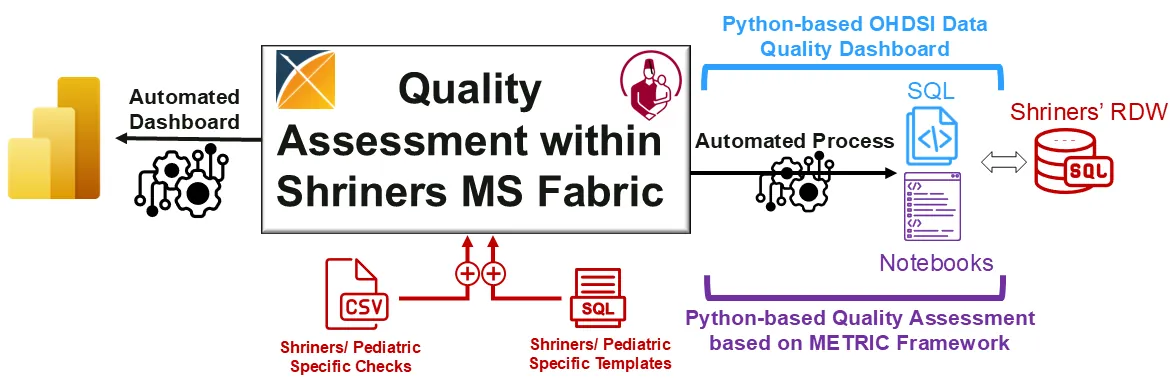
The rapid growth of Artificial Intelligence (AI) in healthcare has sparked interest in Trustworthy AI and AI Implementation Science, both of which are essential for accelerating clinical adoption. However, strict regulations, gaps between research and clinical settings, and challenges in evaluating AI systems continue to hinder real-world implementation. This study presents an AI implementation case study within Shriners Childrens (SC), a large multisite pediatric system, showcasing the modernization of SCs Research Data Warehouse (RDW) to OMOP CDM v5.4 within a secure Microsoft Fabric environment. We introduce a Python-based data quality assessment tool compatible with SCs infrastructure, extending OHDsi's R/Java-based Data Quality Dashboard (DQD) and integrating Trustworthy AI principles using the METRIC framework. This extension enhances data quality evaluation by addressing informative missingness, redundancy, timeliness, and distributional consistency. We also compare systematic and case-specific AI implementation strategies for Craniofacial Microsomia (CFM) using the FHIR standard. Our contributions include a real-world evaluation of AI implementations, integration of Trustworthy AI principles into data quality assessment, and insights into hybrid implementation strategies that blend systematic infrastructure with use-case-driven approaches to advance AI in healthcare.

Protein structure is central to biological function, and enabling multimodal protein models requires joint reasoning over sequence, structure, and function. A key barrier is the lack of principled protein structure tokenizers (PSTs): existing approaches fix token size or rely on continuous vector codebooks, limiting interpretability, multi-scale control, and transfer across architectures. We introduce GeoBPE, a geometry-grounded PST that transforms continuous, noisy, multi-scale backbone conformations into discrete ``sentences'' of geometry while enforcing global constraints. Analogous to byte-pair encoding, GeoBPE generates a hierarchical vocabulary of geometric primitives by iteratively (i) clustering Geo-Pair occurrences with k-medoids to yield a resolution-controllable vocabulary; (ii) quantizing each Geo-Pair to its closest medoid prototype; and (iii) reducing drift through differentiable inverse kinematics that optimizes boundary glue angles under an $\mathrm{SE}(3)$ end-frame loss. GeoBPE offers compression ($>$10x reduction in bits-per-residue at similar distortion rate), data efficiency ($>$10x less training data), and generalization (maintains test/train distortion ratio of $1.0-1.1$). It is architecture-agnostic: (a) its hierarchical vocabulary provides a strong inductive bias for coarsening residue-level embeddings from large PLMs into motif- and protein-level representations, consistently outperforming leading PSTs across $12$ tasks and $24$ test splits; (b) paired with a transformer, GeoBPE supports unconditional backbone generation via language modeling; and (c) tokens align with CATH functional families and support expert-interpretable case studies, offering functional meaning absent in prior PSTs. Code is available at https://github.com/shiningsunnyday/PT-BPE/.
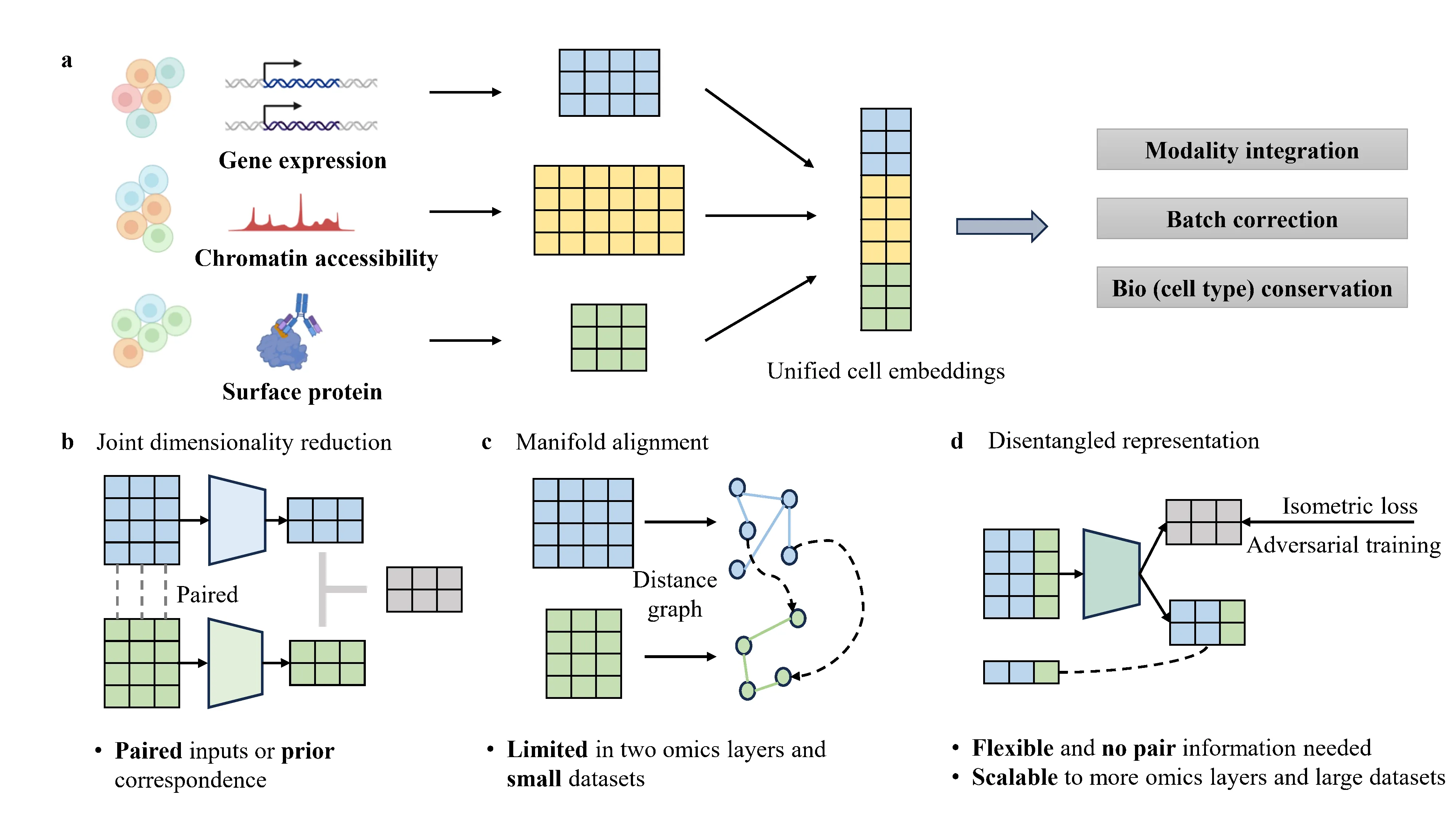
Advances in single-cell sequencing have enabled high-resolution profiling of diverse molecular modalities, while integrating unpaired multi-omics single-cell data remains challenging. Existing approaches either rely on pair information or prior correspondences, or require computing a global pairwise coupling matrix, limiting their scalability and flexibility. In this paper, we introduce a scalable and flexible generative framework called single-cell Multi-omics Regularized Disentangled Representations (scMRDR) for unpaired multi-omics integration. Specifically, we disentangle each cell's latent representations into modality-shared and modality-specific components using a well-designed $β$-VAE architecture, which are augmented with isometric regularization to preserve intra-omics biological heterogeneity, adversarial objective to encourage cross-modal alignment, and masked reconstruction loss strategy to address the issue of missing features across modalities. Our method achieves excellent performance on benchmark datasets in terms of batch correction, modality alignment, and biological signal preservation. Crucially, it scales effectively to large-level datasets and supports integration of more than two omics, offering a powerful and flexible solution for large-scale multi-omics data integration and downstream biological discovery.
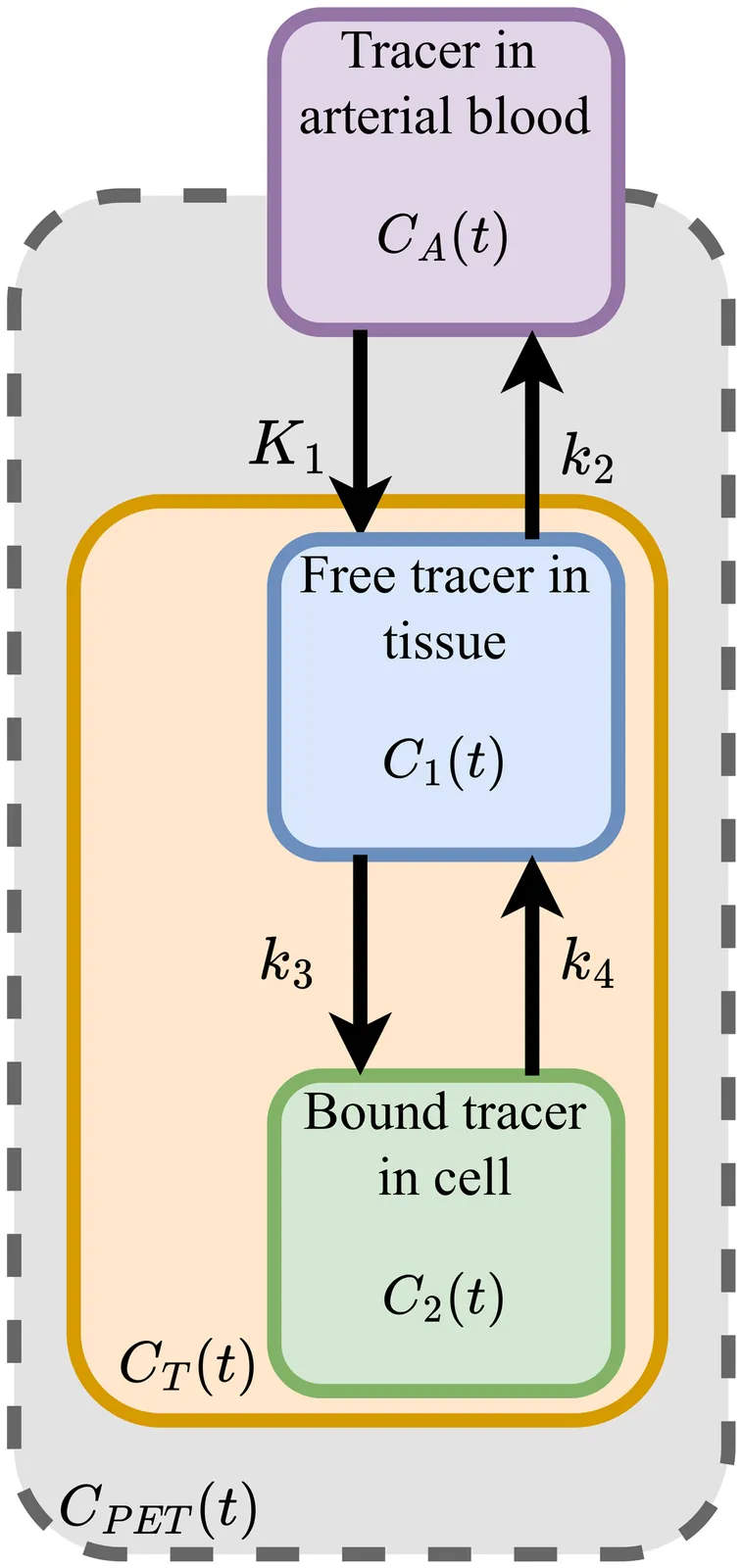
Kinetic modeling enables \textit{in vivo} quantification of tracer uptake and glucose metabolism in [${}^{18}$F]Fluorodeoxyglucose ([${}^{18}$F]FDG) dynamic positron emission tomography (dPET) imaging of mice. However, kinetic modeling requires the accurate determination of the arterial input function (AIF) during imaging, which is time-consuming and invasive. Recent studies have shown the efficacy of using deep learning to directly predict the input function, surpassing established methods such as the image-derived input function (IDIF). In this work, we trained a physics-informed deep learning-based input function prediction model (PIDLIF) to estimate the AIF directly from the PET images, incorporating a kinetic modeling loss during training. The proposed method uses a two-tissue compartment model over two regions, the myocardium and brain of the mice, and is trained on a dataset of 70 [${}^{18}$F]FDG dPET images of mice accompanied by the measured AIF during imaging. The proposed method had comparable performance to the network without a physics-informed loss, and when sudden movement causing blurring in the images was simulated, the PIDLIF model maintained high performance in severe cases of image degradation. The proposed physics-informed method exhibits an improved robustness that is promoted by physically constraining the problem, enforcing consistency for out-of-distribution samples. In conclusion, the PIDLIF model offers insight into the effects of leveraging physiological distribution mechanics in mice to guide a deep learning-based AIF prediction network in images with severe degradation as a result of blurring due to movement during imaging.
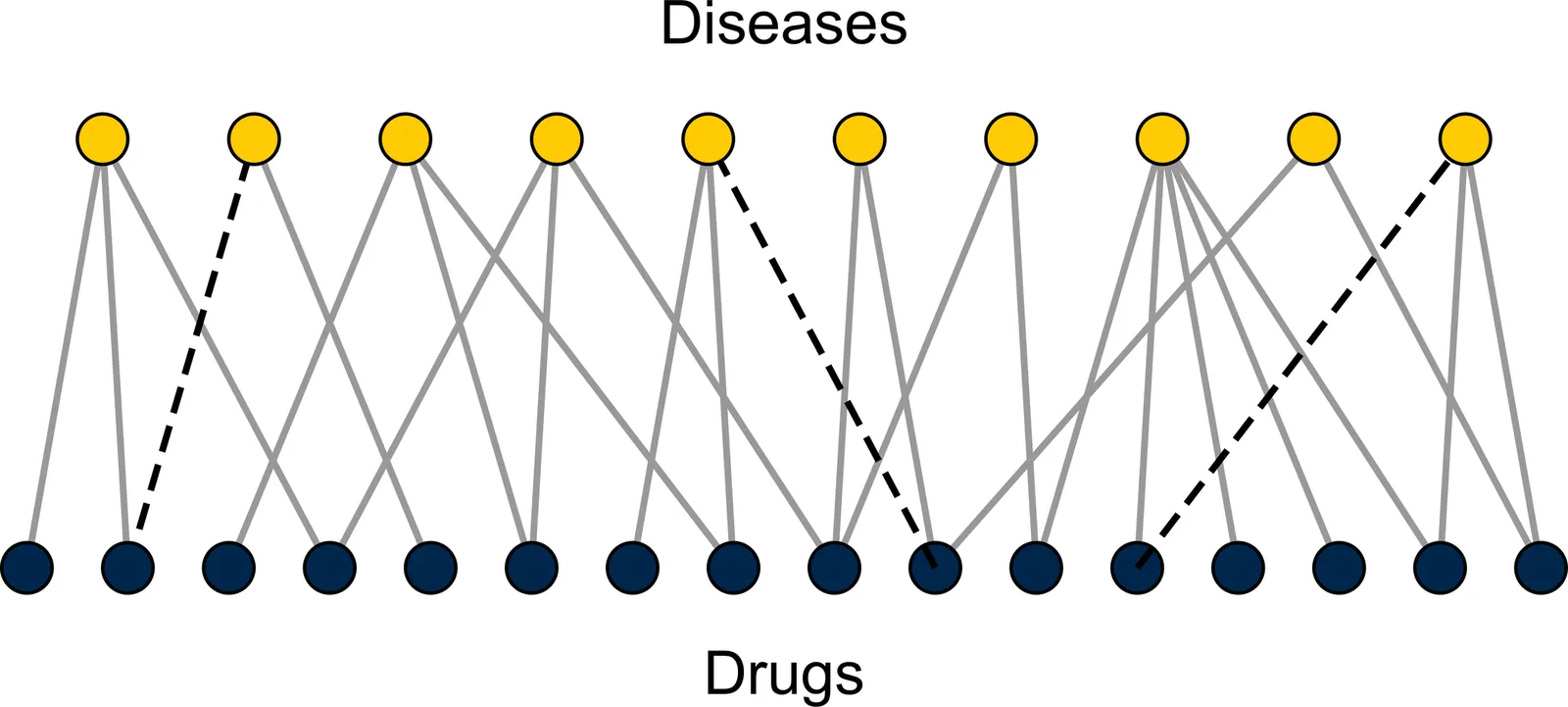
Repurposing existing drugs to treat new diseases is a cost-effective alternative to de novo drug development, but there are millions of potential drug-disease combinations to be considered with only a small fraction being viable. In silico predictions of drug-disease associations can be invaluable for reducing the size of the search space. In this work we present a novel network of drugs and the diseases they treat, compiled using a combination of existing textual and machine-readable databases, natural-language processing tools, and hand curation, and analyze it using network-based link prediction methods to identify potential drug-disease combinations. We measure the efficacy of these methods using cross-validation tests and find that several methods, particularly those based on graph embedding and network model fitting, achieve impressive prediction performance, significantly better than previous approaches, with area under the ROC curve above 0.95 and average precision almost a thousand times better than chance.
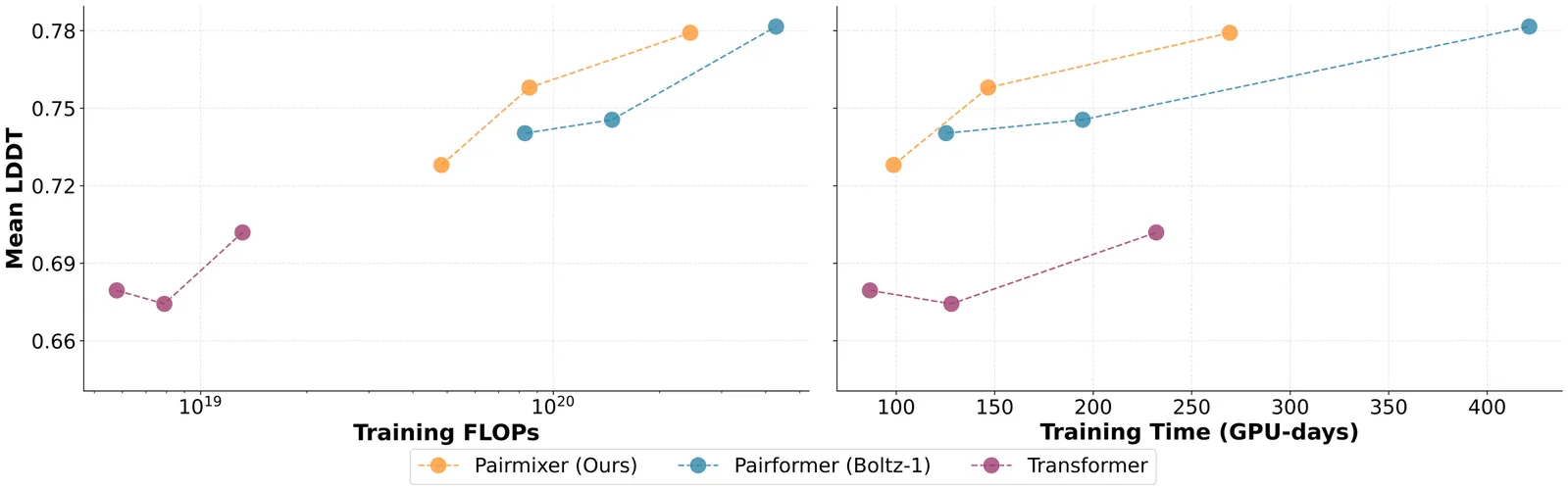
AlphaFold has transformed protein structure prediction, but emerging applications such as virtual ligand screening, proteome-wide folding, and de novo binder design demand predictions at a massive scale, where runtime and memory costs become prohibitive. A major bottleneck lies in the Pairformer backbone of AlphaFold3-style models, which relies on computationally expensive triangular primitives-especially triangle attention-for pairwise reasoning. We introduce Pairmixer, a streamlined alternative that eliminates triangle attention while preserving higher-order geometric reasoning capabilities that are critical for structure prediction. Pairmixer substantially improves computational efficiency, matching state-of-the-art structure predictors across folding and docking benchmarks, delivering up to 4x faster inference on long sequences while reducing training cost by 34%. Its efficiency alleviates the computational burden of downstream applications such as modeling large protein complexes, high-throughput ligand and binder screening, and hallucination-based design. Within BoltzDesign, for example, Pairmixer delivers over 2x faster sampling and scales to sequences ~30% longer than the memory limits of Pairformer. Code is available at https://github.com/genesistherapeutics/pairmixer.
Designing protein sequences that fold into a target three-dimensional structure, known as the inverse folding problem, is central to protein engineering but remains challenging due to the vast sequence space and the importance of local structural constraints. Existing deep learning approaches achieve strong recovery rates, yet they lack explicit mechanisms to reuse fine-grained structure-sequence patterns that are conserved across natural proteins. We present PRISM, a multimodal retrieval-augmented generation framework for inverse folding that retrieves fine-grained representations of potential motifs from known proteins and integrates them with a hybrid self-cross attention decoder. PRISM is formulated as a latent-variable probabilistic model and implemented with an efficient approximation, combining theoretical grounding with practical scalability. Across five benchmarks (CATH-4.2, TS50, TS500, CAMEO 2022, and the PDB date split), PRISM establishes new state of the art in both perplexity and amino acid recovery, while also improving foldability metrics (RMSD, TM-score, pLDDT), demonstrating that fine-grained multimodal retrieval is a powerful and efficient paradigm for protein sequence design.
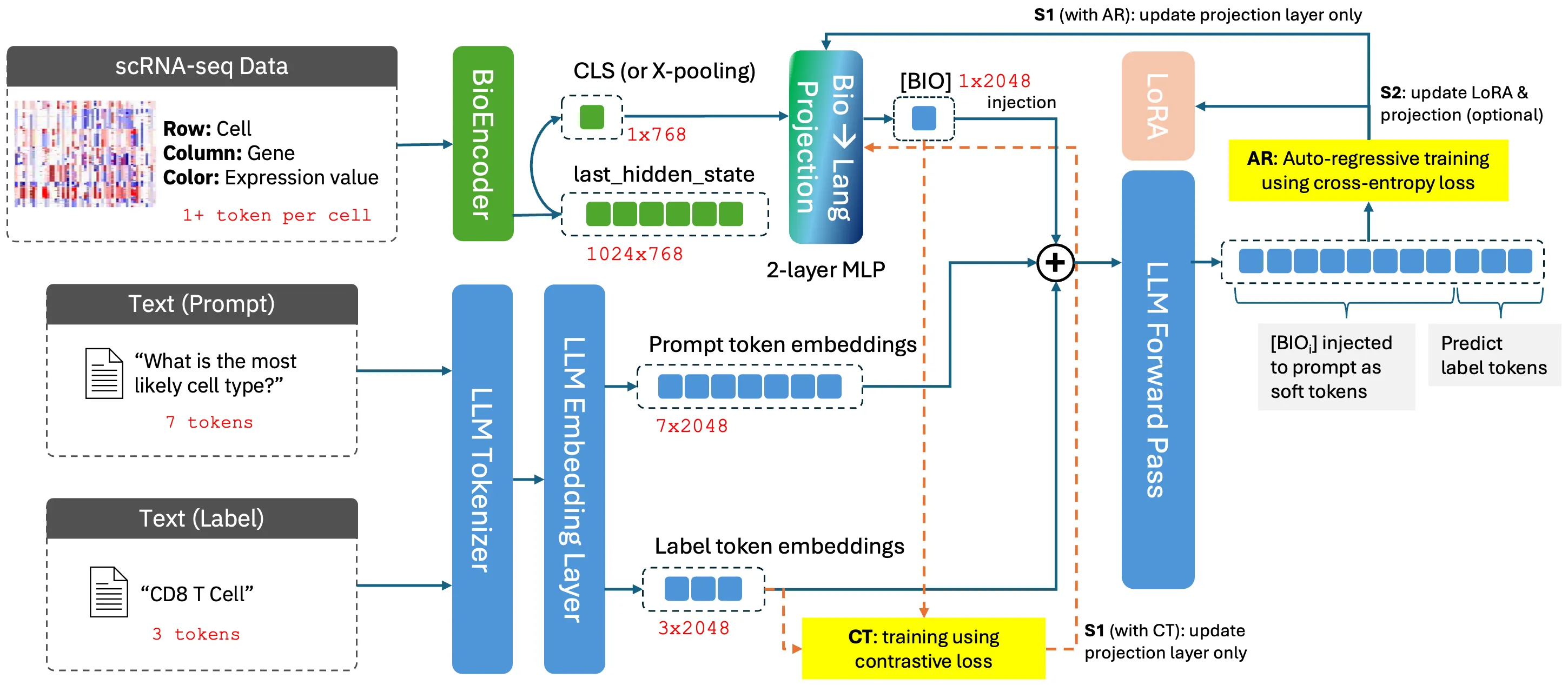
Recent advances in large language models (LLMs) and biomedical foundation models (BioFMs) have achieved strong results in biological text reasoning, molecular modeling, and single-cell analysis, yet they remain siloed in disjoint embedding spaces, limiting cross-modal reasoning. We present BIOVERSE (Biomedical Vector Embedding Realignment for Semantic Engagement), a two-stage approach that adapts pretrained BioFMs as modality encoders and aligns them with LLMs through lightweight, modality-specific projection layers. The approach first aligns each modality to a shared LLM space through independently trained projections, allowing them to interoperate naturally, and then applies standard instruction tuning with multi-modal data to bring them together for downstream reasoning. By unifying raw biomedical data with knowledge embedded in LLMs, the approach enables zero-shot annotation, cross-modal question answering, and interactive, explainable dialogue. Across tasks spanning cell-type annotation, molecular description, and protein function reasoning, compact BIOVERSE configurations surpass larger LLM baselines while enabling richer, generative outputs than existing BioFMs, establishing a foundation for principled multi-modal biomedical reasoning.
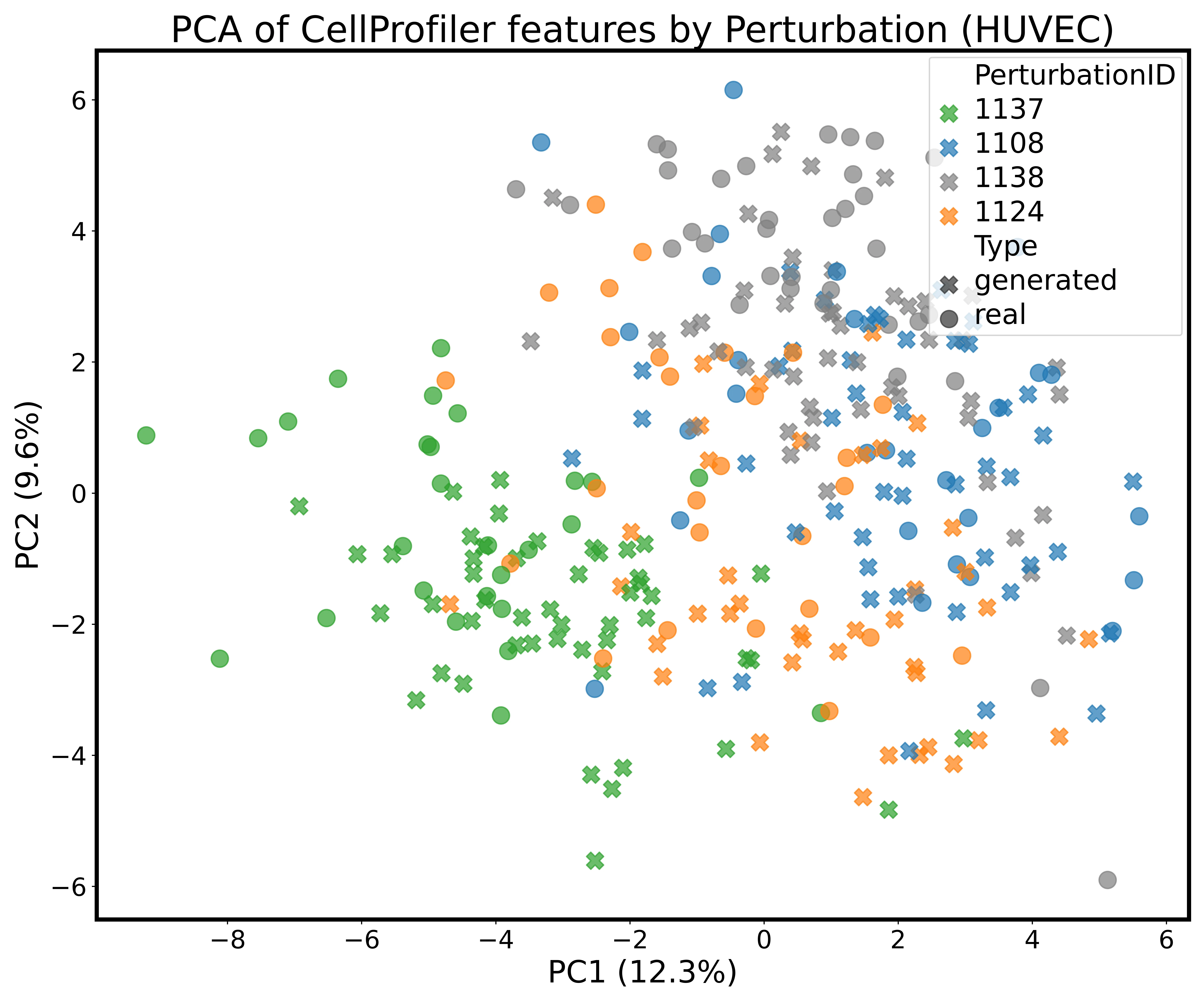
Simulating in silico cellular responses to interventions is a promising direction to accelerate high-content image-based assays, critical for advancing drug discovery and gene editing. To support this, we introduce MorphGen, a state-of-the-art diffusion-based generative model for fluorescent microscopy that enables controllable generation across multiple cell types and perturbations. To capture biologically meaningful patterns consistent with known cellular morphologies, MorphGen is trained with an alignment loss to match its representations to the phenotypic embeddings of OpenPhenom, a state-of-the-art biological foundation model. Unlike prior approaches that compress multichannel stains into RGB images -- thus sacrificing organelle-specific detail -- MorphGen generates the complete set of fluorescent channels jointly, preserving per-organelle structures and enabling a fine-grained morphological analysis that is essential for biological interpretation. We demonstrate biological consistency with real images via CellProfiler features, and MorphGen attains an FID score over 35% lower than the prior state-of-the-art MorphoDiff, which only generates RGB images for a single cell type. Code is available at https://github.com/czi-ai/MorphGen.
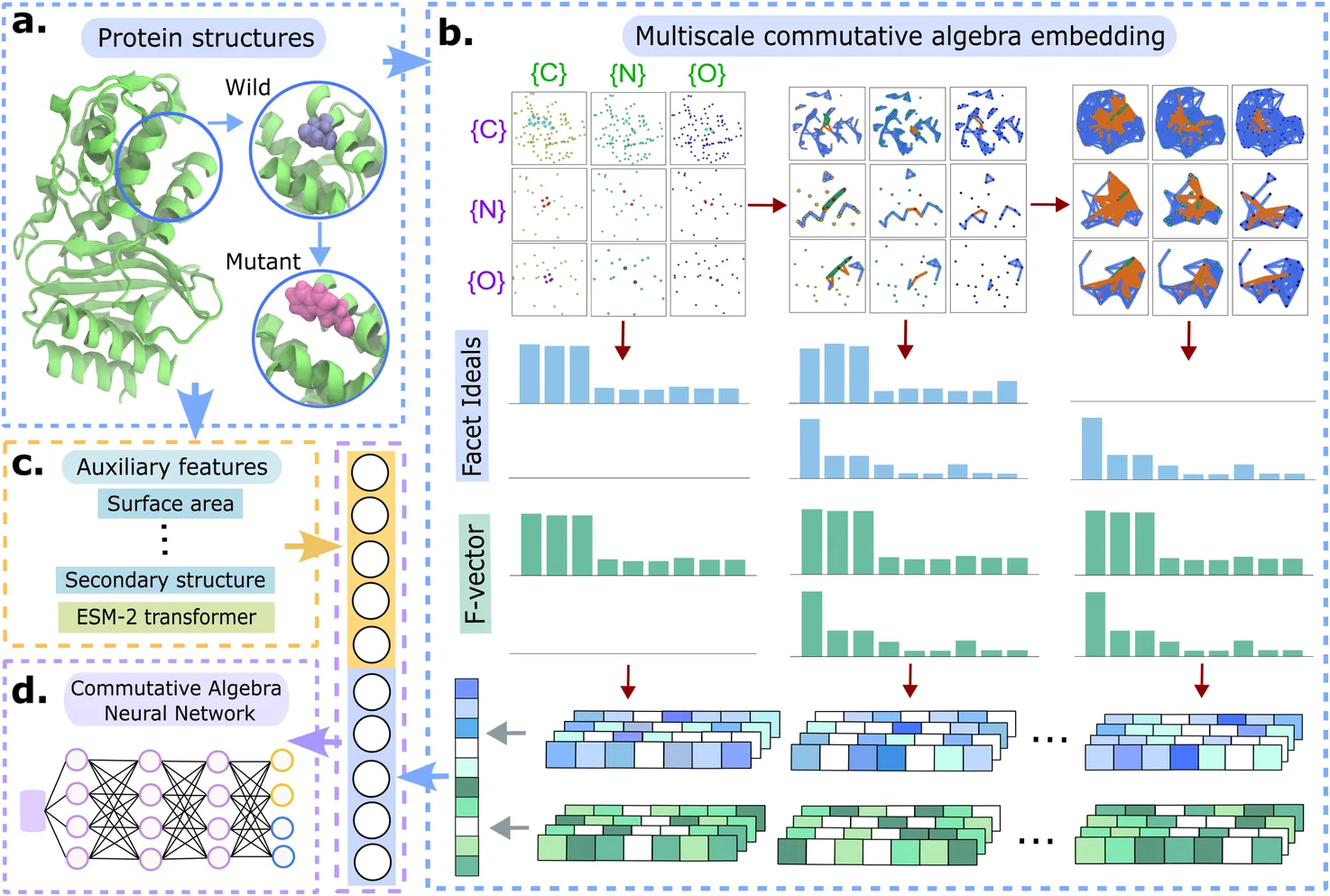
Genetic mutations can disrupt protein structure, stability, and solubility, contributing to a wide range of diseases. Existing predictive models often lack interpretability and fail to integrate physical and chemical interactions critical to molecular mechanisms. Moreover, current approaches treat disease association, stability changes, and solubility alterations as separate tasks, limiting model generalizability. In this study, we introduce a unified framework based on multiscale commutative algebra to capture intrinsic physical and chemical interactions for the first time. Leveraging Persistent Stanley-Reisner Theory, we extract multiscale algebraic invariants to build a Commutative Algebra neural Network (CANet). Integrated with transformer features and auxiliary physical features, we apply CANet to tackle three key domains for the first time: disease-associated mutations, mutation-induced protein stability changes, and solubility changes upon mutations. Across six benchmark tasks, CANet and its gradient boosting tree counterpart, CATree, consistently attain state-of-the-art performance, achieving up to 7.5% improvement in predictive accuracy. Our approach offers multiscale, mechanistic, interpretable,and generalizable models for predicting disease-mutation associations.

We introduce AbBiBench (Antibody Binding Benchmarking), a benchmarking framework for antibody binding affinity maturation and design. Unlike previous strategies that evaluate antibodies in isolation, typically by comparing them to natural sequences with metrics such as amino acid recovery rate or structural RMSD, AbBiBench instead treats the antibody-antigen (Ab-Ag) complex as the fundamental unit. It evaluates an antibody design's binding potential by measuring how well a protein model scores the full Ab-Ag complex. We first curate, standardize, and share more than 184,500 experimental measurements of antibody mutants across 14 antibodies and 9 antigens-including influenza, lysozyme, HER2, VEGF, integrin, Ang2, and SARS-CoV-2-covering both heavy-chain and light-chain mutations. Using these datasets, we systematically compare 15 protein models including masked language models, autoregressive language models, inverse folding models, diffusion-based generative models, and geometric graph models by comparing the correlation between model likelihood and experimental affinity values. Additionally, to demonstrate AbBiBench's generative utility, we apply it to antibody F045-092 in order to introduce binding to influenza H1N1. We sample new antibody variants with the top-performing models, rank them by the structural integrity and biophysical properties of the Ab-Ag complex, and assess them with in vitro ELISA binding assays. Our findings show that structure-conditioned inverse folding models outperform others in both affinity correlation and generation tasks. Overall, AbBiBench provides a unified, biologically grounded evaluation framework to facilitate the development of more effective, function-aware antibody design models.

Mechanistic, multicellular, agent-based models are commonly used to investigate tissue, organ, and organism-scale biology at single-cell resolution. The Cellular-Potts Model (CPM) is a powerful and popular framework for developing and interrogating these models. CPMs become computationally expensive at large space- and time- scales making application and investigation of developed models difficult. Surrogate models may allow for the accelerated evaluation of CPMs of complex biological systems. However, the stochastic nature of these models means each set of parameters may give rise to different model configurations, complicating surrogate model development. In this work, we leverage denoising diffusion probabilistic models to train a generative AI surrogate of a CPM used to investigate in vitro vasculogenesis. We describe the use of an image classifier to learn the characteristics that define unique areas of a 2-dimensional parameter space. We then apply this classifier to aid in surrogate model selection and verification. Our CPM model surrogate generates model configurations 20,000 timesteps ahead of a reference configuration and demonstrates approximately a 22x reduction in computational time as compared to native code execution. Our work represents a step towards the implementation of DDPMs to develop digital twins of stochastic biological systems.
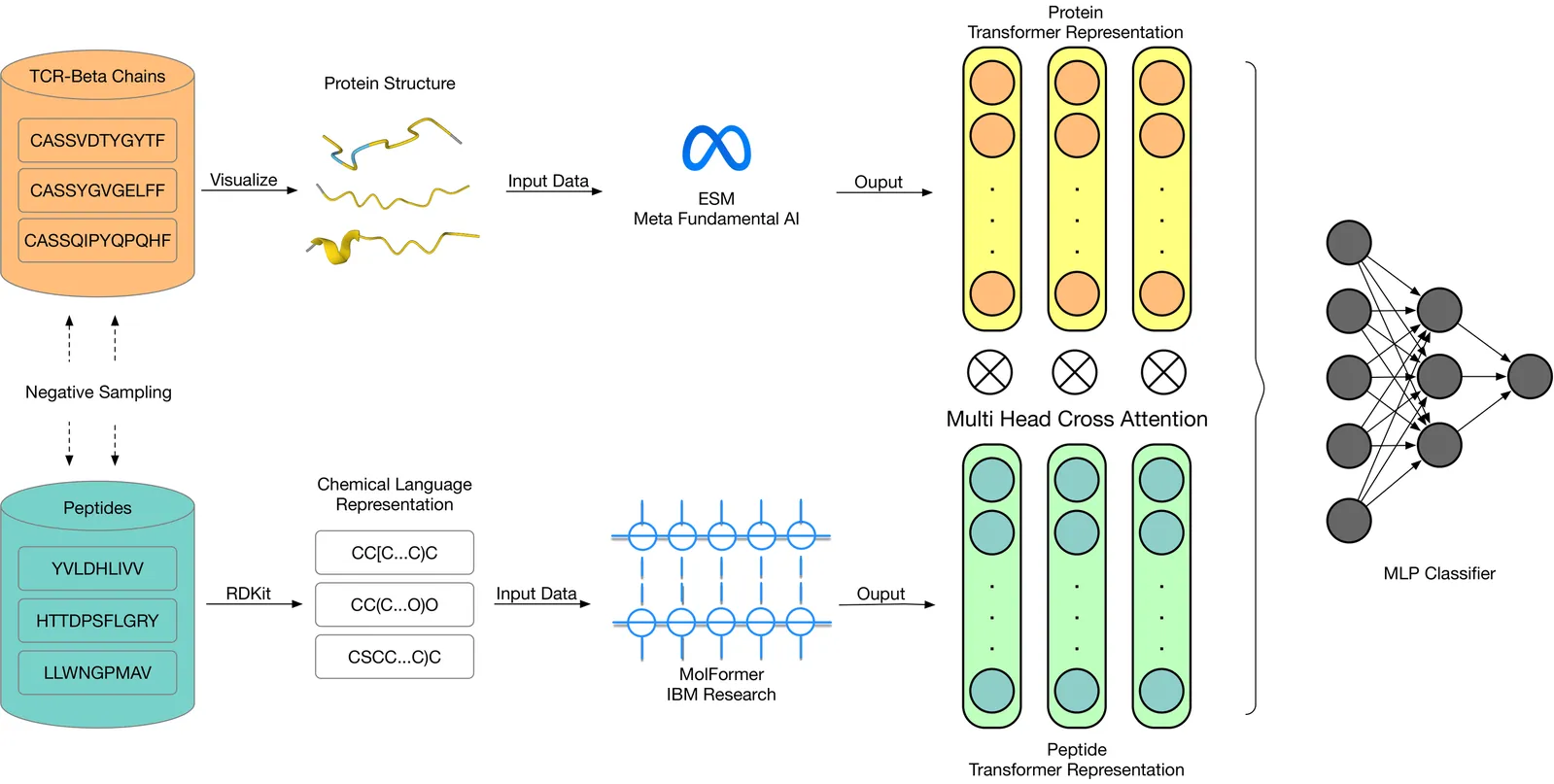
Understanding the binding specificity between T-cell receptors (TCRs) and peptide-major histocompatibility complexes (pMHCs) is central to immunotherapy and vaccine development. However, current predictive models struggle with generalization, especially in data-scarce settings and when faced with novel epitopes. We present LANTERN (Large lAnguage model-powered TCR-Enhanced Recognition Network), a deep learning framework that combines large-scale protein language models with chemical representations of peptides. By encoding TCR \b{eta}-chain sequences using ESM-1b and transforming peptide sequences into SMILES strings processed by MolFormer, LANTERN captures rich biological and chemical features critical for TCR-peptide recognition. Through extensive benchmarking against existing models such as ChemBERTa, TITAN, and NetTCR, LANTERN demonstrates superior performance, particularly in zero-shot and few-shot learning scenarios. Our model also benefits from a robust negative sampling strategy and shows significant clustering improvements via embedding analysis. These results highlight the potential of LANTERN to advance TCR-pMHC binding prediction and support the development of personalized immunotherapies.
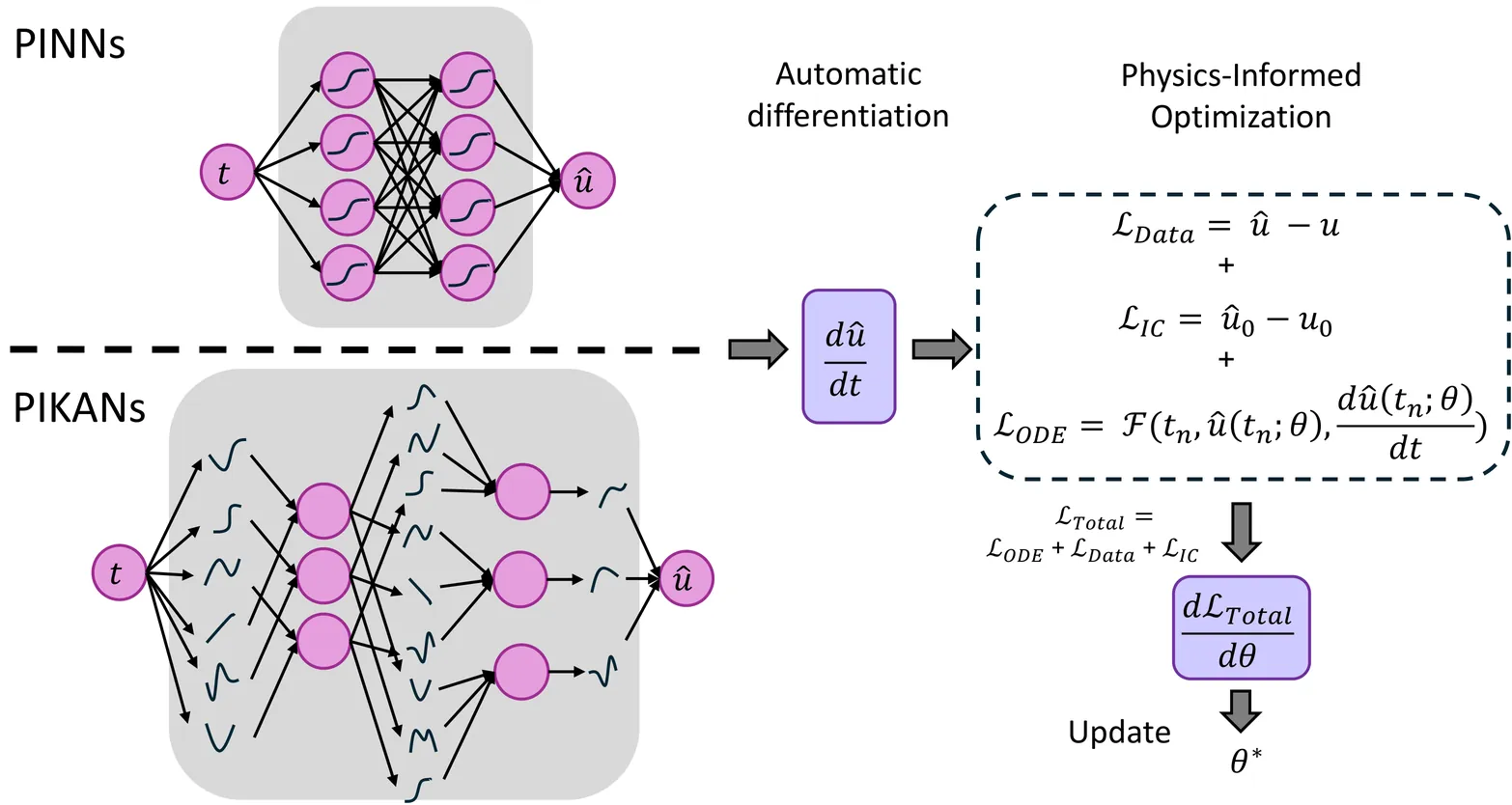
Physics-Informed Kolmogorov-Arnold Networks (PIKANs) are gaining attention as an effective counterpart to the original multilayer perceptron-based Physics-Informed Neural Networks (PINNs). Both representation models can address inverse problems and facilitate gray-box system identification. However, a comprehensive understanding of their performance in terms of accuracy and speed remains underexplored. In particular, we introduce a modified PIKAN architecture, tanh-cPIKAN, which is based on Chebyshev polynomials for parametrization of the univariate functions with an extra nonlinearity for enhanced performance. We then present a systematic investigation of how choices of the optimizer, representation, and training configuration influence the performance of PINNs and PIKANs in the context of systems pharmacology modeling. We benchmark a wide range of first-order, second-order, and hybrid optimizers, including various learning rate schedulers. We use the new Optax library to identify the most effective combinations for learning gray-boxes under ill-posed, non-unique, and data-sparse conditions. We examine the influence of model architecture (MLP vs. KAN), numerical precision (single vs. double), the need for warm-up phases for second-order methods, and sensitivity to the initial learning rate. We also assess the optimizer scalability for larger models and analyze the trade-offs introduced by JAX in terms of computational efficiency and numerical accuracy. Using two representative systems pharmacology case studies - a pharmacokinetics model and a chemotherapy drug-response model - we offer practical guidance on selecting optimizers and representation models/architectures for robust and efficient gray-box discovery. Our findings provide actionable insights for improving the training of physics-informed networks in biomedical applications and beyond.
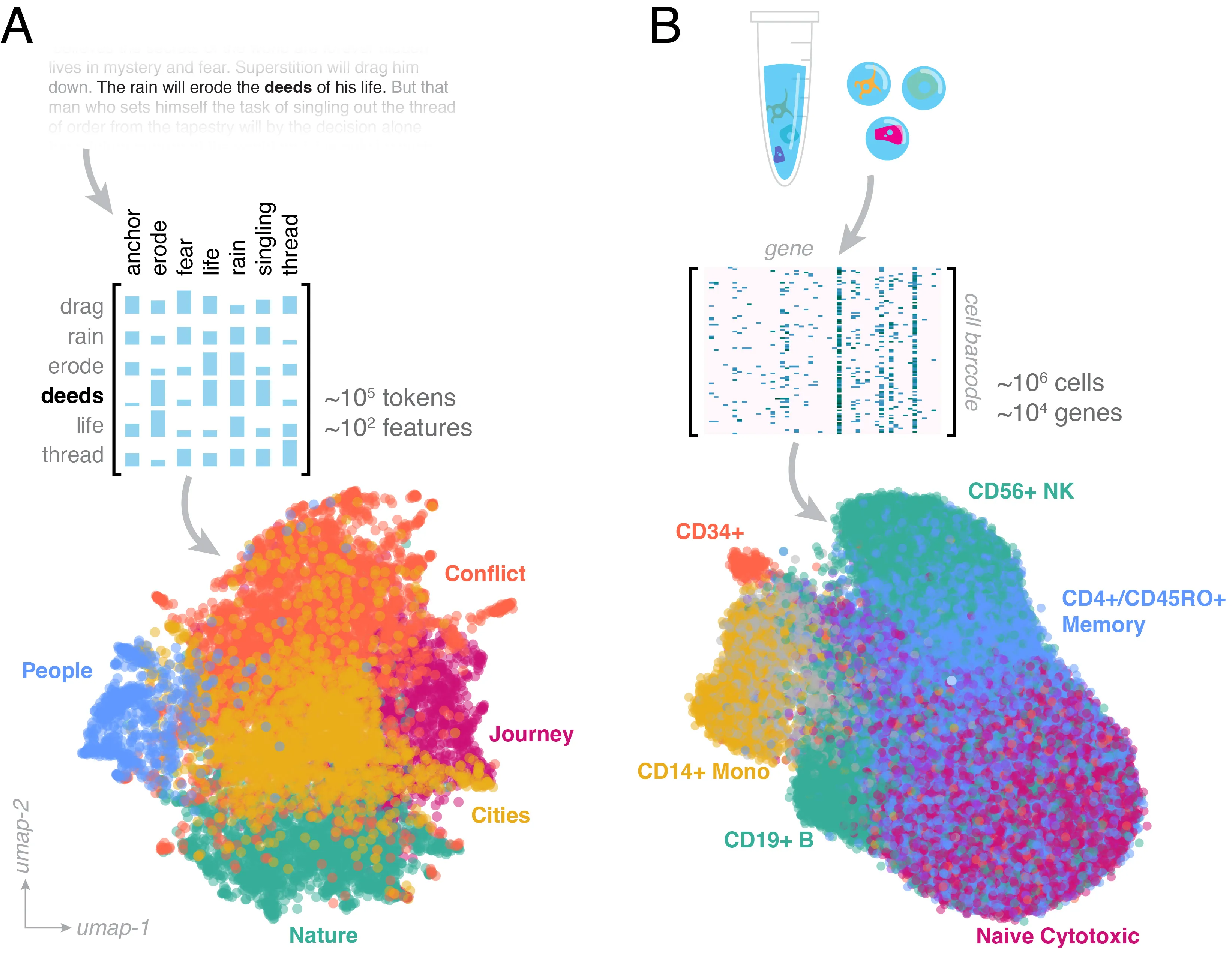
Single-cell sequencing technology maps cells to a high-dimensional space encoding their internal activity. Recently-proposed virtual cell models extend this concept, enriching cells' representations based on patterns learned from pretraining on vast cell atlases. This review explores how advances in understanding the structure of natural language embeddings informs ongoing efforts to analyze single-cell datasets. Both fields process unstructured data by partitioning datasets into tokens embedded within a high-dimensional vector space. We discuss how the context of tokens influences the geometry of embedding space, and how low-dimensional manifolds shape this space's robustness and interpretation. We highlight how new developments in foundation models for language, such as interpretability probes and in-context reasoning, can inform efforts to construct cell atlases and train virtual cell models.
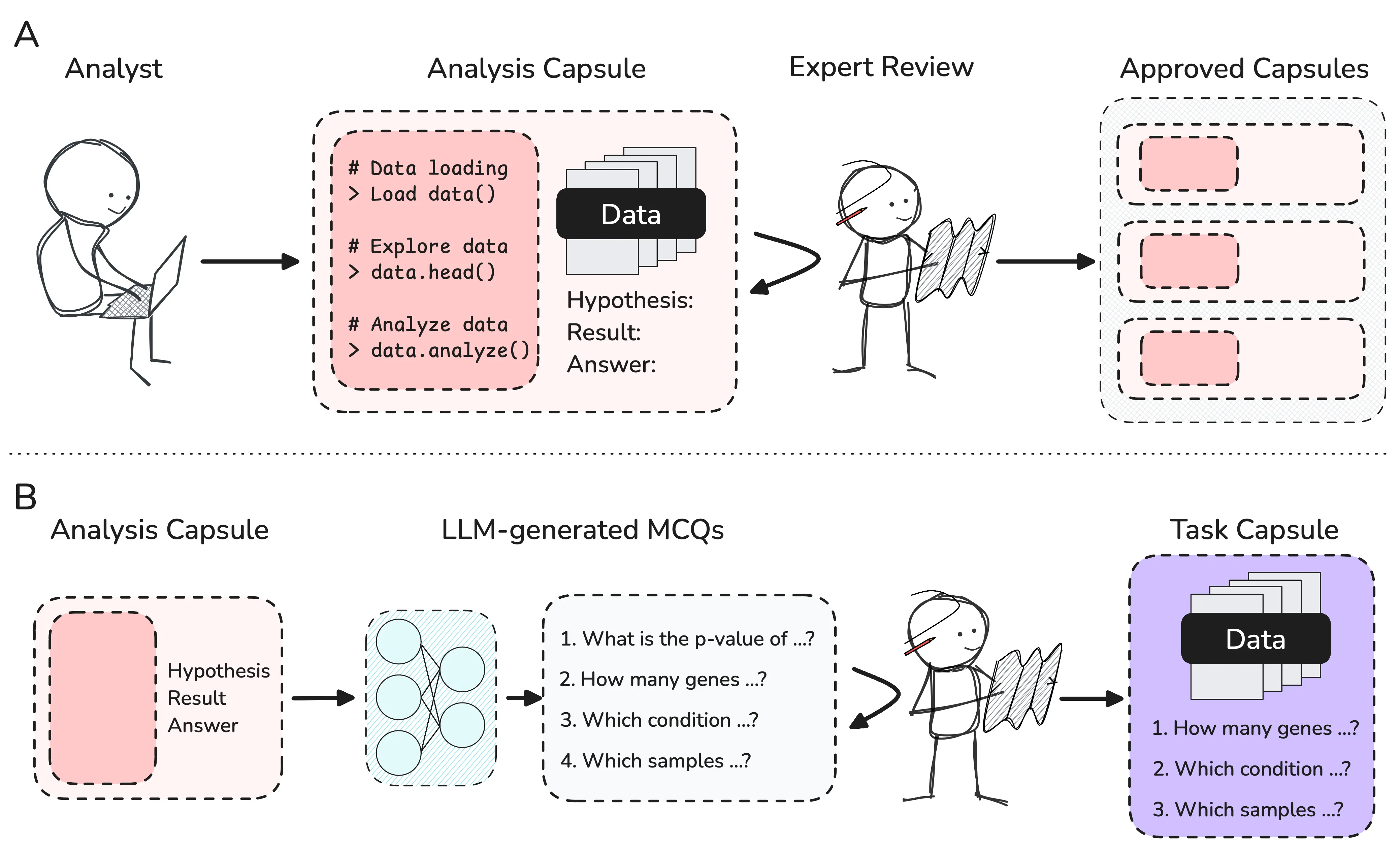
Large Language Models (LLMs) and LLM-based agents show great promise in accelerating scientific research. Existing benchmarks for measuring this potential and guiding future development continue to evolve from pure recall and rote knowledge tasks, towards more practical work such as literature review and experimental planning. Bioinformatics is a domain where fully autonomous AI-driven discovery may be near, but no extensive benchmarks for measuring progress have been introduced to date. We therefore present the Bioinformatics Benchmark (BixBench), a dataset comprising over 50 real-world scenarios of practical biological data analysis with nearly 300 associated open-answer questions designed to measure the ability of LLM-based agents to explore biological datasets, perform long, multi-step analytical trajectories, and interpret the nuanced results of those analyses. We evaluate the performance of two frontier LLMs (GPT-4o and Claude 3.5 Sonnet) using a custom agent framework we open source. We find that even the latest frontier models only achieve 17% accuracy in the open-answer regime, and no better than random in a multiple-choice setting. By exposing the current limitations of frontier models, we hope BixBench can spur the development of agents capable of conducting rigorous bioinformatic analysis and accelerate scientific discovery.
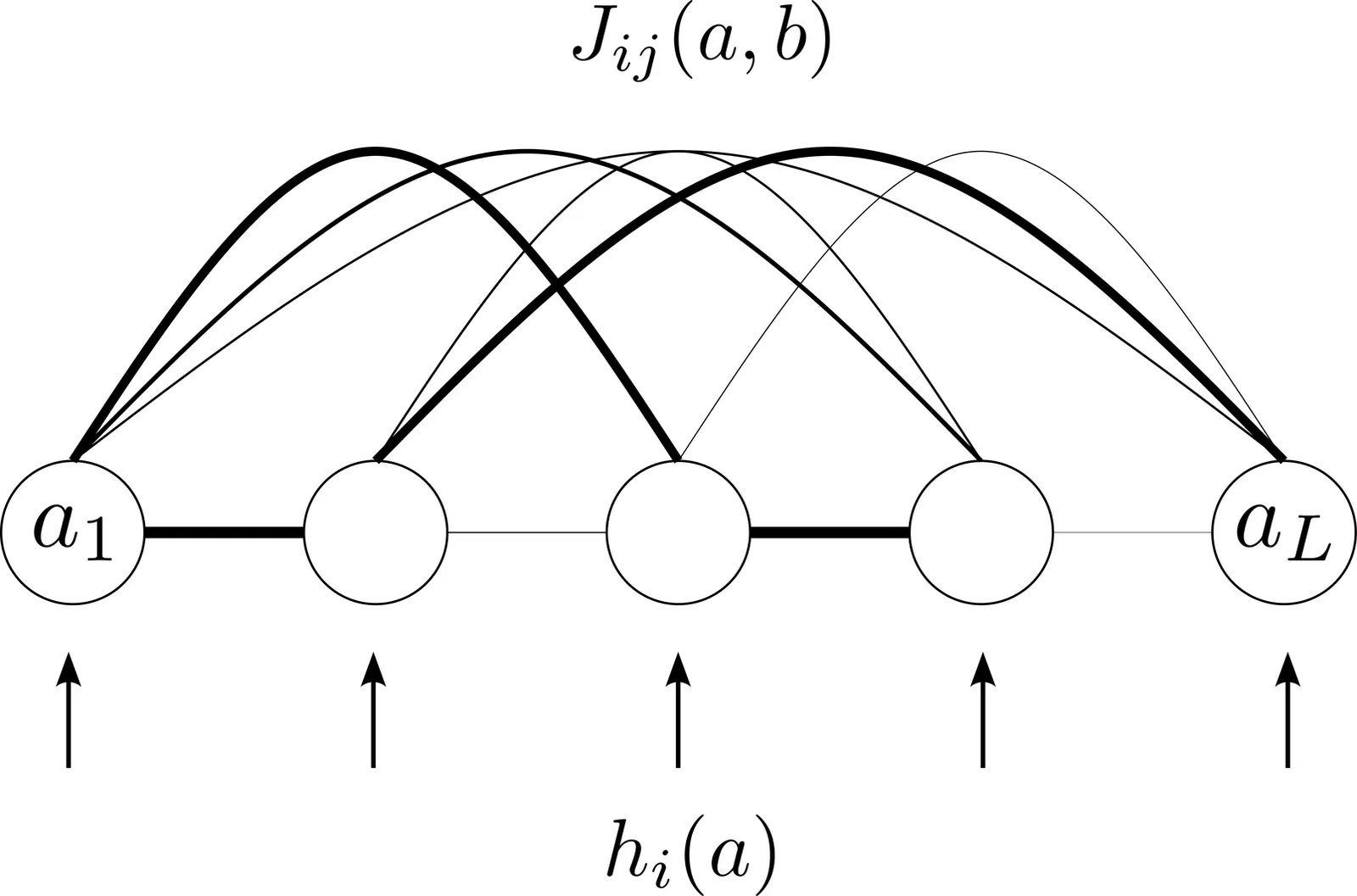
In this methods article, we provide a flexible but easy-to-use implementation of Direct Coupling Analysis (DCA) based on Boltzmann machine learning, together with a tutorial on how to use it. The package \texttt{adabmDCA 2.0} is available in different programming languages (C++, Julia, Python) usable on different architectures (single-core and multi-core CPU, GPU) using a common front-end interface. In addition to several learning protocols for dense and sparse generative DCA models, it allows to directly address common downstream tasks like residue-residue contact prediction, mutational-effect prediction, scoring of sequence libraries and generation of artificial sequences for sequence design. It is readily applicable to protein and RNA sequence data.
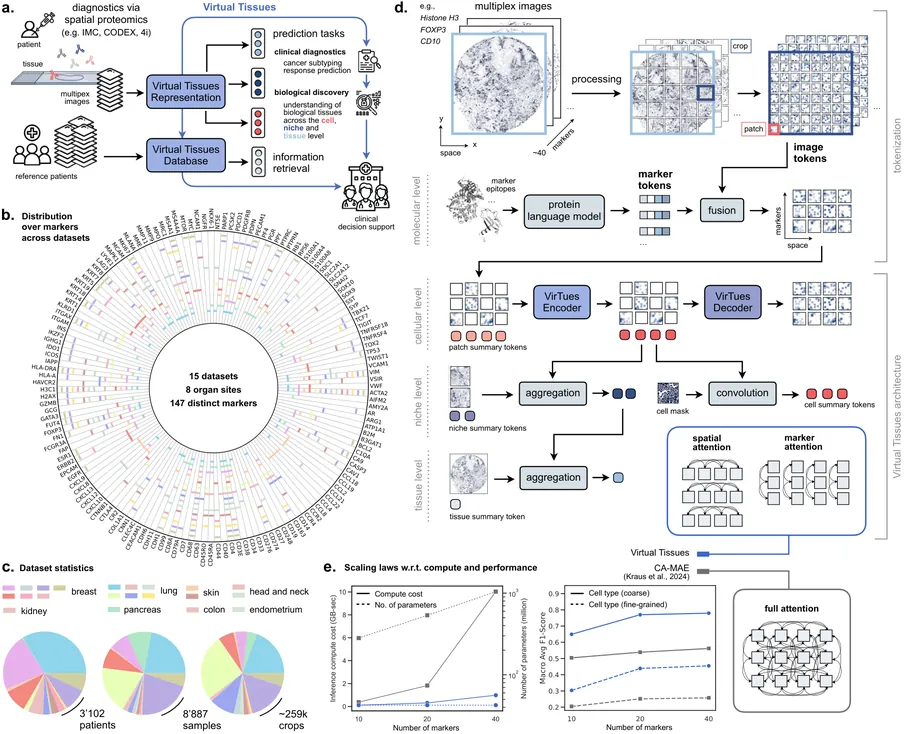
Spatial proteomics technologies have transformed our understanding of complex tissue architecture in cancer but present unique challenges for computational analysis. Each study uses a different marker panel and protocol, and most methods are tailored to single cohorts, which limits knowledge transfer and robust biomarker discovery. Here we present Virtual Tissues (VirTues), a general-purpose foundation model for spatial proteomics that learns marker-aware, multi-scale representations of proteins, cells, niches and tissues directly from multiplex imaging data. From a single pretrained backbone, VirTues supports marker reconstruction, cell typing and niche annotation, spatial biomarker discovery, and patient stratification, including zero-shot annotation across heterogeneous panels and datasets. In triple-negative breast cancer, VirTues-derived biomarkers predict anti-PD-L1 chemo-immunotherapy response and stratify disease-free survival in an independent cohort, outperforming state-of-the-art biomarkers derived from the same datasets and current clinical stratification schemes.
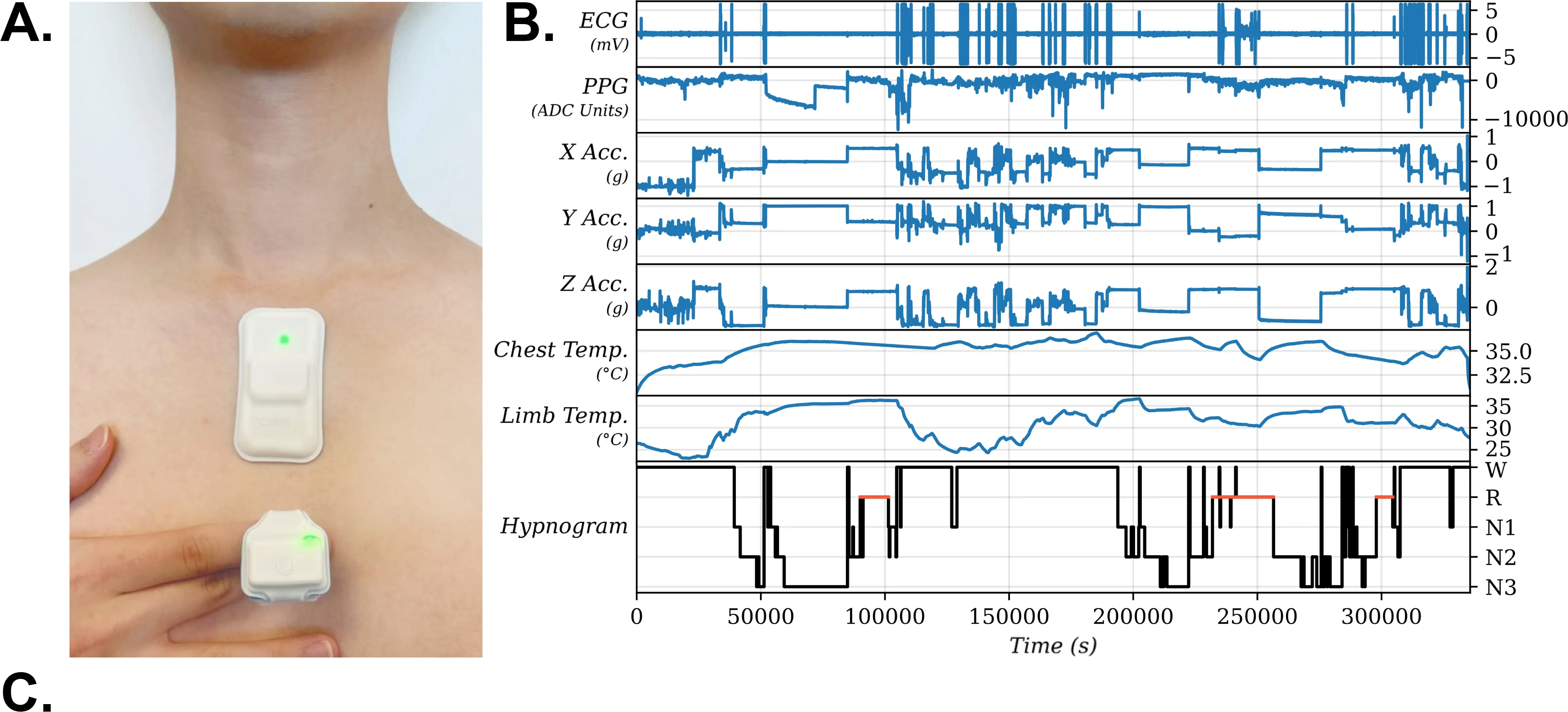
Study Objectives: We investigate a Mamba-based deep learning approach for sleep staging on signals from ANNE One (Sibel Health, Evanston, IL), a non-intrusive dual-module wireless wearable system measuring chest electrocardiography (ECG), triaxial accelerometry, and chest temperature, and finger photoplethysmography and finger temperature. Methods: We obtained wearable sensor recordings from 357 adults undergoing concurrent polysomnography (PSG) at a tertiary care sleep lab. Each PSG recording was manually scored and these annotations served as ground truth labels for training and evaluation of our models. PSG and wearable sensor data were automatically aligned using their ECG channels with manual confirmation by visual inspection. We trained a Mamba-based recurrent neural network architecture on these recordings. Ensembling of model variants with similar architectures was performed. Results: After ensembling, the model attains a 3-class (wake, non rapid eye movement [NREM] sleep, rapid eye movement [REM] sleep) balanced accuracy of 84.02%, F1 score of 84.23%, Cohen's $κ$ of 72.89%, and a Matthews correlation coefficient (MCC) score of 73.00%; a 4-class (wake, light NREM [N1/N2], deep NREM [N3], REM) balanced accuracy of 75.30%, F1 score of 74.10%, Cohen's $κ$ of 61.51%, and MCC score of 61.95%; a 5-class (wake, N1, N2, N3, REM) balanced accuracy of 65.11%, F1 score of 66.15%, Cohen's $κ$ of 53.23%, MCC score of 54.38%. Conclusions: Our Mamba-based deep learning model can successfully infer major sleep stages from the ANNE One, a wearable system without electroencephalography (EEG), and can be applied to data from adults attending a tertiary care sleep clinic.
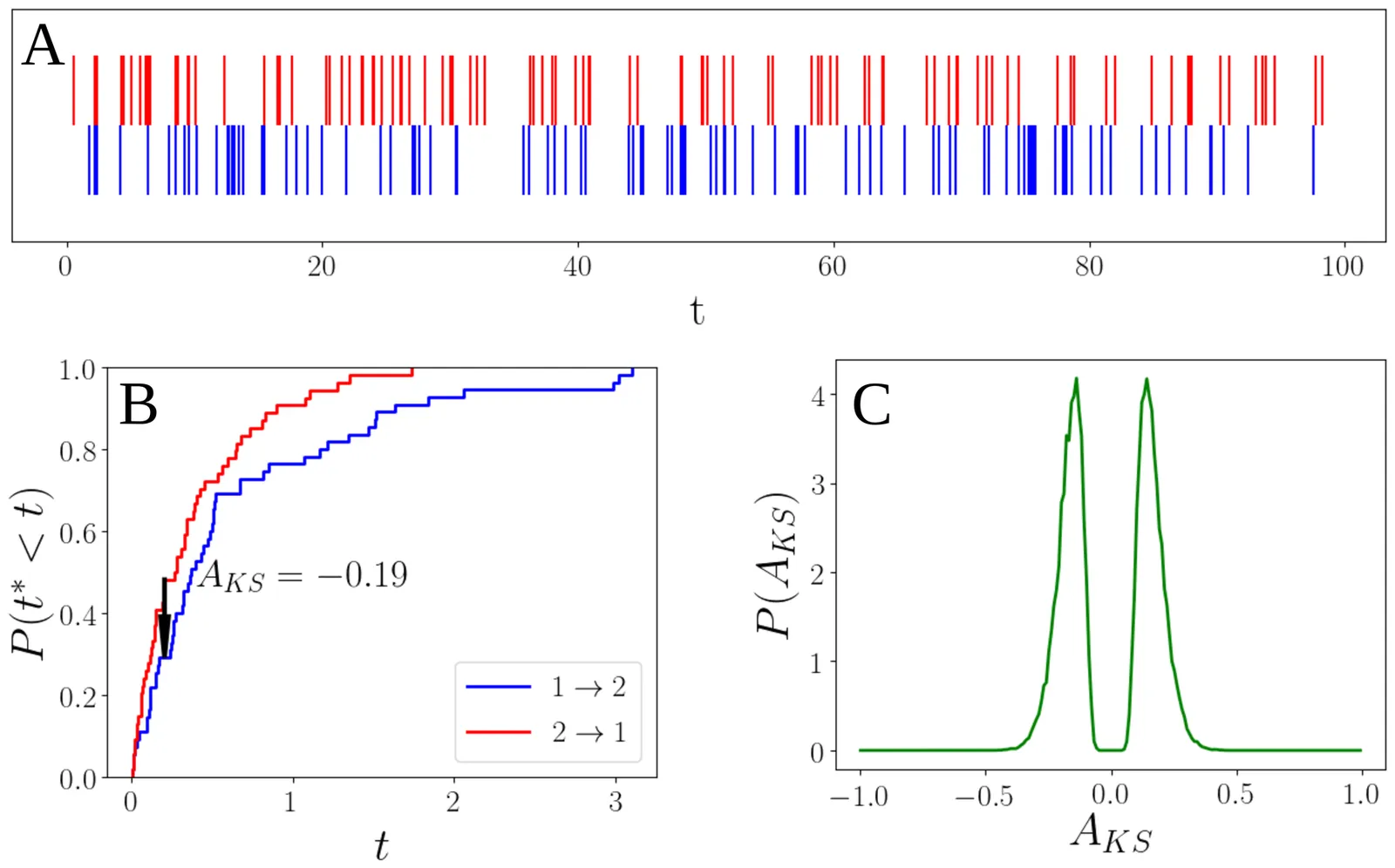
Social interactions are fundamental in animal groups, including humans, and can take various forms, such as competition, cooperation, or kinship. Understanding these interactions in marine environments has been historically challenging due to data collection difficulties. However, advancements in acoustic telemetry now enable remote analysis of such behaviors. This study proposes a method to derive leader-follower networks from presence data collected by a single acoustic receiver at a specific location. Using the Kolmogorov-Smirnov distance, the method analyzes lag times between consecutive presences of individuals to infer directed relationships. Tested on simulated data, it was then applied to detection data from acoustically tagged reef manta rays (\textit{Mobula~alfredi}) frequenting a known site. Results revealed temporal patterns, including circadian rhythms and burst-like behavior with power-law distributed time gaps between presences. The inferred leader-follower network highlighted key behavioral patterns: females followed males more often than expected, males showed stronger but fewer associations with specific females, and smaller individuals followed others less consistently than larger ones. These findings align with ecological insights, revealing structured social interactions and providing a novel framework for studying marine animal behavior through network theory.
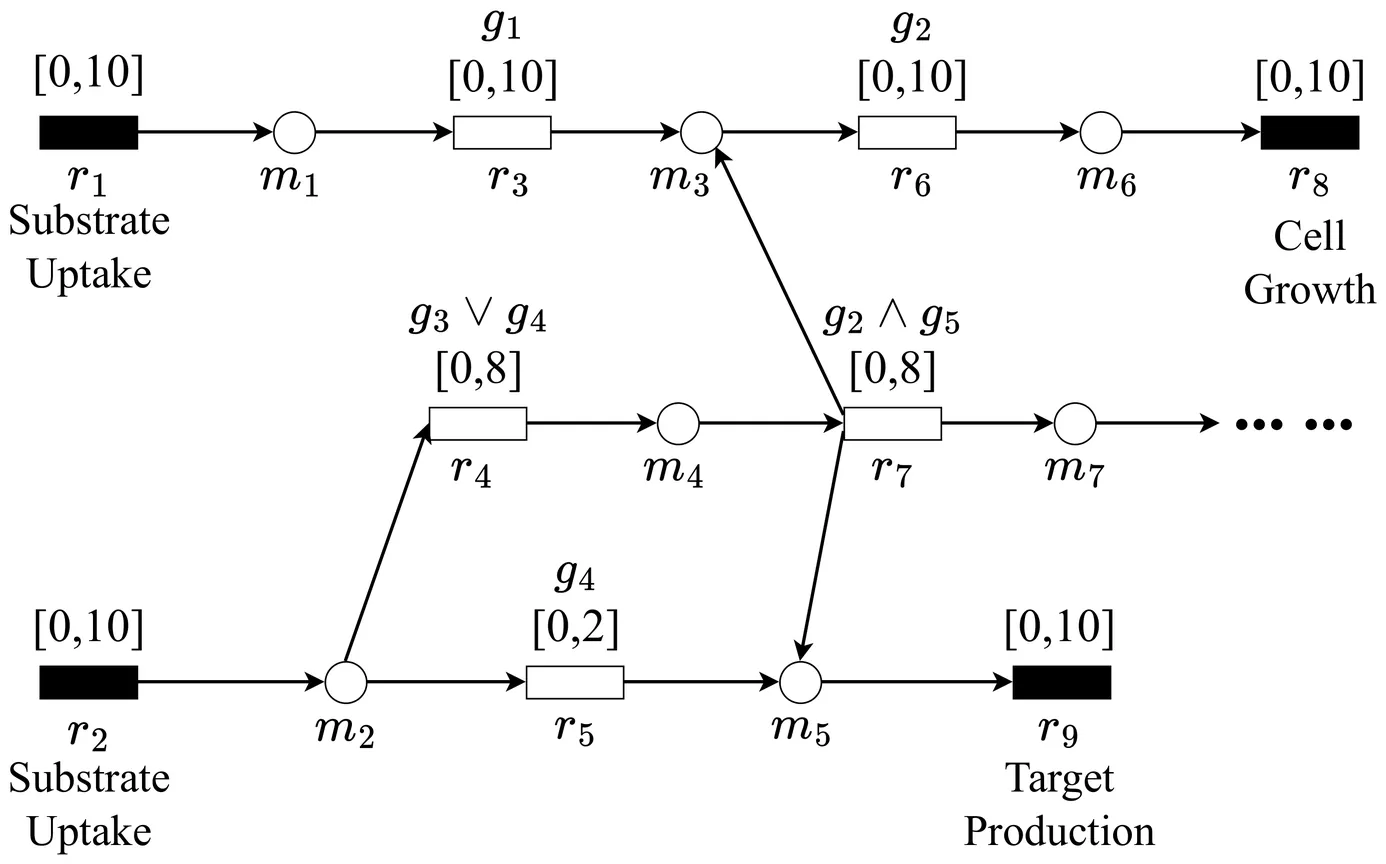
When simulating metabolite productions with genome-scale constraint-based metabolic models, gene deletion strategies are necessary to achieve growth-coupled production, which means cell growth and target metabolite production occur simultaneously. Since obtaining gene deletion strategies for large genome-scale models suffers from significant computational time, it is necessary to develop methods to mitigate this computational burden. In this study, we introduce a novel framework for computing gene deletion strategies. The proposed framework first mines related databases to extract prior information about gene deletions for growth-coupled production. It then integrates the extracted information with downstream algorithms to narrow down the algorithmic search space, resulting in highly efficient calculations on genome-scale models. Computational experiment results demonstrated that our framework can compute stoichiometrically feasible gene deletion strategies for numerous target metabolites, showcasing a noteworthy improvement in computational efficiency. Specifically, our framework achieves an average 6.1-fold acceleration in computational speed compared to existing methods while maintaining a respectable success rate. The source code of DBgDel with examples are available on https://github.com/MetNetComp/DBgDel.