Genomics
arXiv:q-bio.GN
DNA/RNA sequencing and analysis, gene finding, genome assembly and annotation, transcriptomics.
Looking for a broader view? This category is part of:
DNA/RNA sequencing and analysis, gene finding, genome assembly and annotation, transcriptomics.
Looking for a broader view? This category is part of:
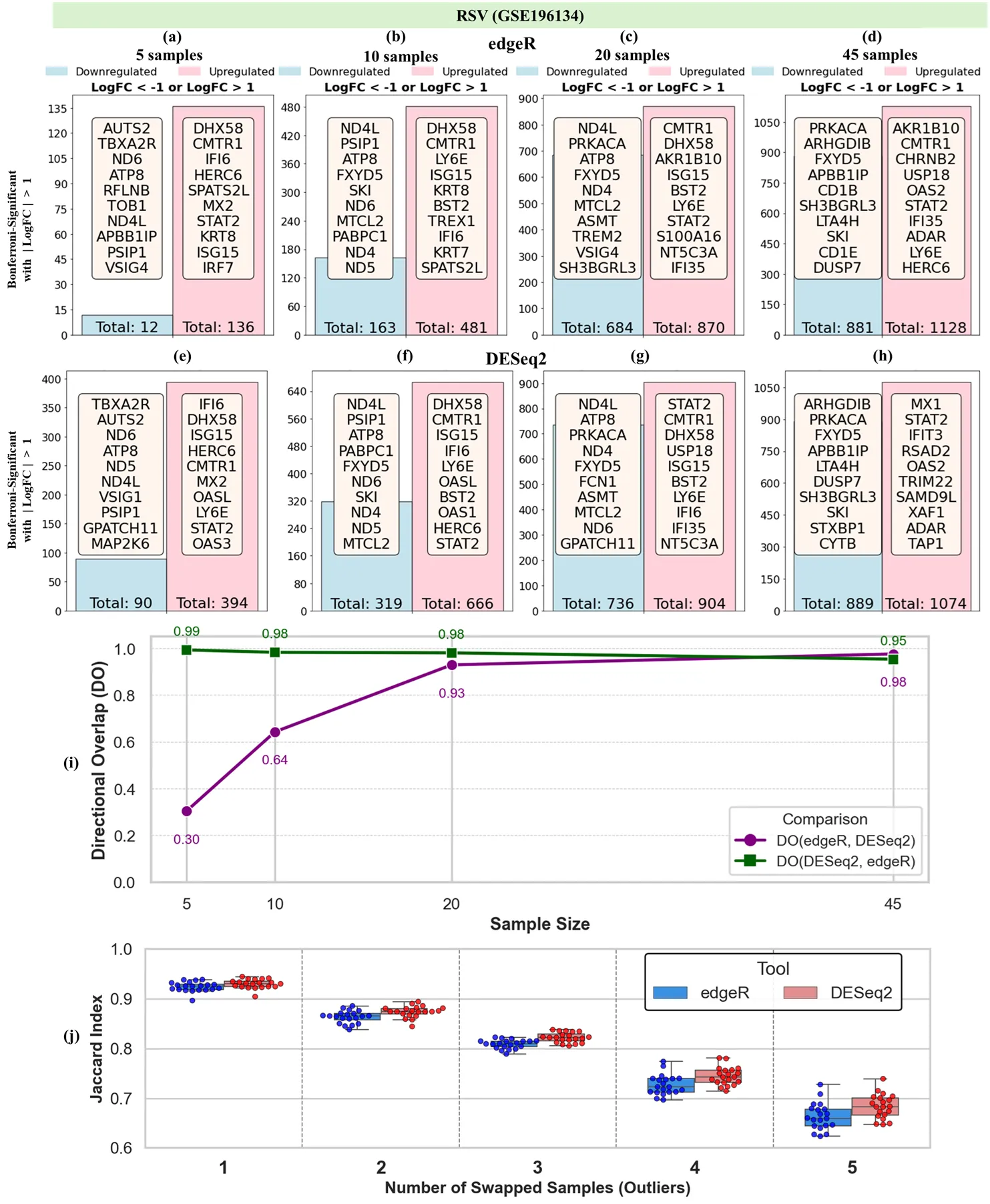
Differential gene expression (DGE) analysis is foundational to transcriptomic research, yet tool selection can substantially influence results. This study presents a comprehensive comparison of two widely used DGE tools, edgeR and DESeq2, using real and semi-simulated bulk RNA-Seq datasets spanning viral, bacterial, and fibrotic conditions. We evaluated tool performance across three key dimensions: (1) sensitivity to sample size and robustness to outliers; (2) classification performance of uniquely identified gene sets within the discovery dataset; and (3) generalizability of tool-specific gene sets across independent studies. First, both tools showed similar responses to simulated outliers, with Jaccard similarity between the DEG sets from perturbed and original (unperturbed) data decreasing as more outliers were added. Second, classification models trained on tool-specific genes showed that edgeR achieved higher F1 scores in 9 of 13 contrasts and more frequently reached perfect or near-perfect precision. Dolan-More performance profiles further indicated that edgeR maintained performance closer to optimal across a greater proportion of datasets. Third, in cross-study validation using four independent SARS-CoV-2 datasets, gene sets uniquely identified by edgeR yielded higher AUC, precision, and recall in classifying samples from held-out datasets. This pattern was consistent across folds, with some test cases achieving perfect separation using edgeR-specific genes. In contrast, DESeq2-specific genes showed lower and more variable performance across studies. Overall, our findings highlight that while DESeq2 may identify more DEGs even under stringent significance conditions, edgeR yields more robust and generalizable gene sets for downstream classification and cross-study replication, which underscores key trade-offs in tool selection for transcriptomic analyses.
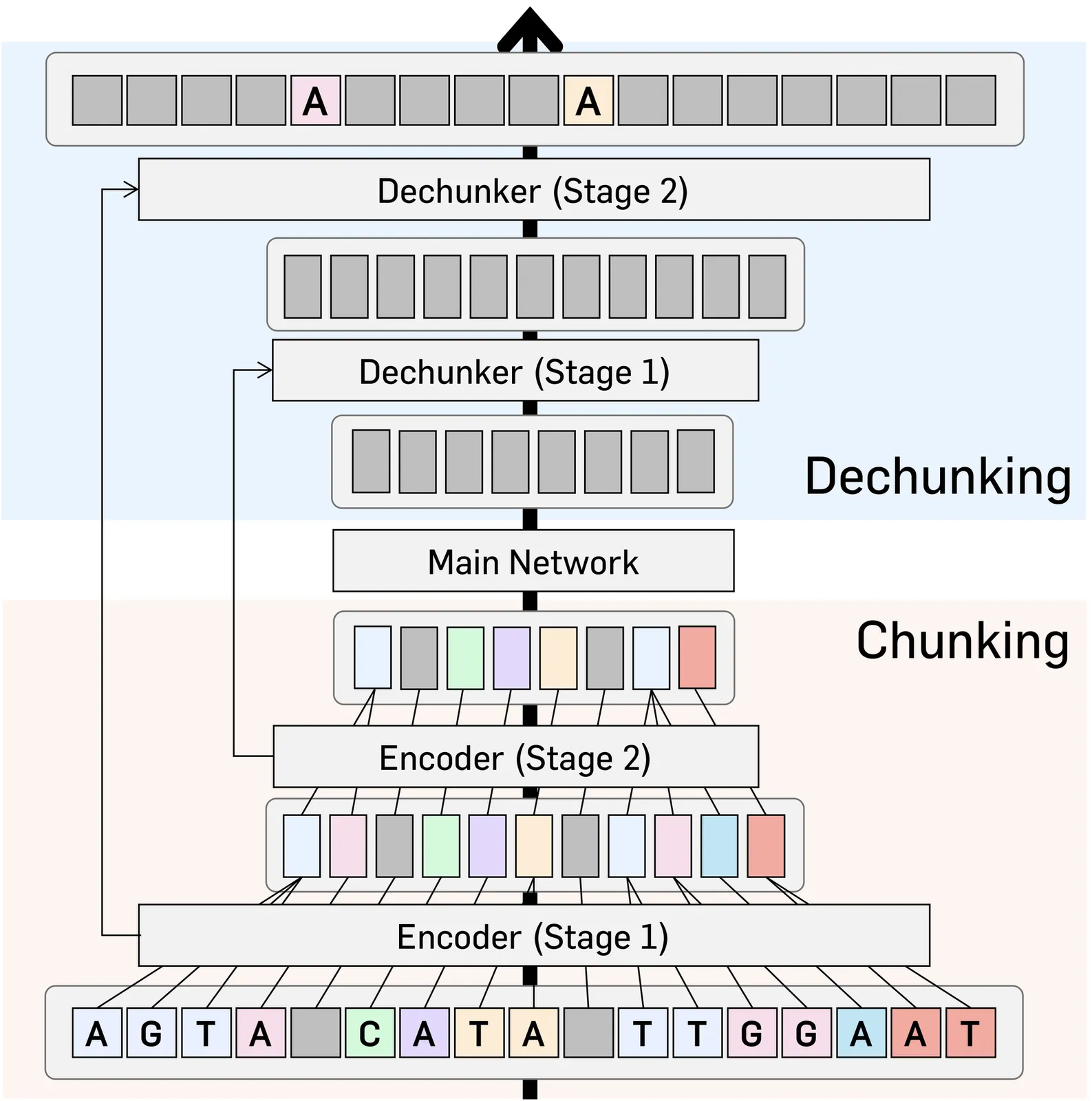
DNA language models have emerged as powerful tools for decoding the complex language of DNA sequences. However, the performance of these models is heavily affected by their tokenization strategy, i.e., a method used to parse DNA sequences into a shorter sequence of chunks. In this work, we propose DNACHUNKER, which integrates a learnable dynamic DNA tokenization mechanism and is trained as a masked language model. Adopting the dynamic chunking procedure proposed by H-Net, our model learns to segment sequences into variable-length chunks. This dynamic chunking offers two key advantages: it's resilient to shifts and mutations in the DNA, and it allocates more detail to important functional areas. We demonstrate the performance of DNACHUNKER by training it on the human reference genome (HG38) and testing it on the Nucleotide Transformer and Genomic benchmarks. Further ablative experiments reveal that DNACHUNKER learns tokenization that grasps biological grammar and uses smaller chunks to preserve detail in important functional elements such as promoters and exons, while using larger chunks for repetitive, redundant regions.
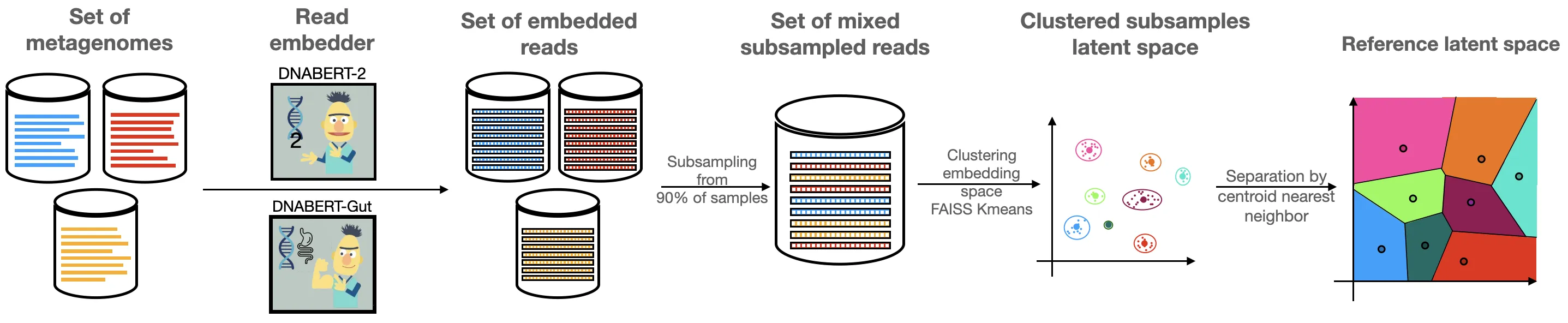
Metagenomic disease prediction commonly relies on species abundance tables derived from large, incomplete reference catalogs, constraining resolution and discarding valuable information contained in DNA reads. To overcome these limitations, we introduce MetagenBERT, a Transformer based framework that produces end to end metagenome embeddings directly from raw DNA sequences, without taxonomic or functional annotations. Reads are embedded using foundational genomic language models (DNABERT2 and the microbiome specialized DNABERTMS), then aggregated through a scalable clustering strategy based on FAISS accelerated KMeans. Each metagenome is represented as a cluster abundance vector summarizing the distribution of its embedded reads. We evaluate this approach on five benchmark gut microbiome datasets (Cirrhosis, T2D, Obesity, IBD, CRC). MetagenBERT achieves competitive or superior AUC performance relative to species abundance baselines across most tasks. Concatenating both representations further improves prediction, demonstrating complementarity between taxonomic and embedding derived signals. Clustering remains robust when applied to as little as 10% of reads, highlighting substantial redundancy in metagenomes and enabling major computational gains. We additionally introduce MetagenBERT Glob Mcardis, a cross cohort variant trained on the large, phenotypically diverse MetaCardis cohort and transferred to other datasets, retaining predictive signal including for unseen phenotypes, indicating the feasibility of a foundation model for metagenome representation. Robustness analyses (PERMANOVA, PERMDISP, entropy) show consistent separation of different states across subsamples. Overall, MetagenBERT provides a scalable, annotation free representation of metagenomes pointing toward future phenotype aware generalization across heterogeneous cohorts and sequencing technologies.
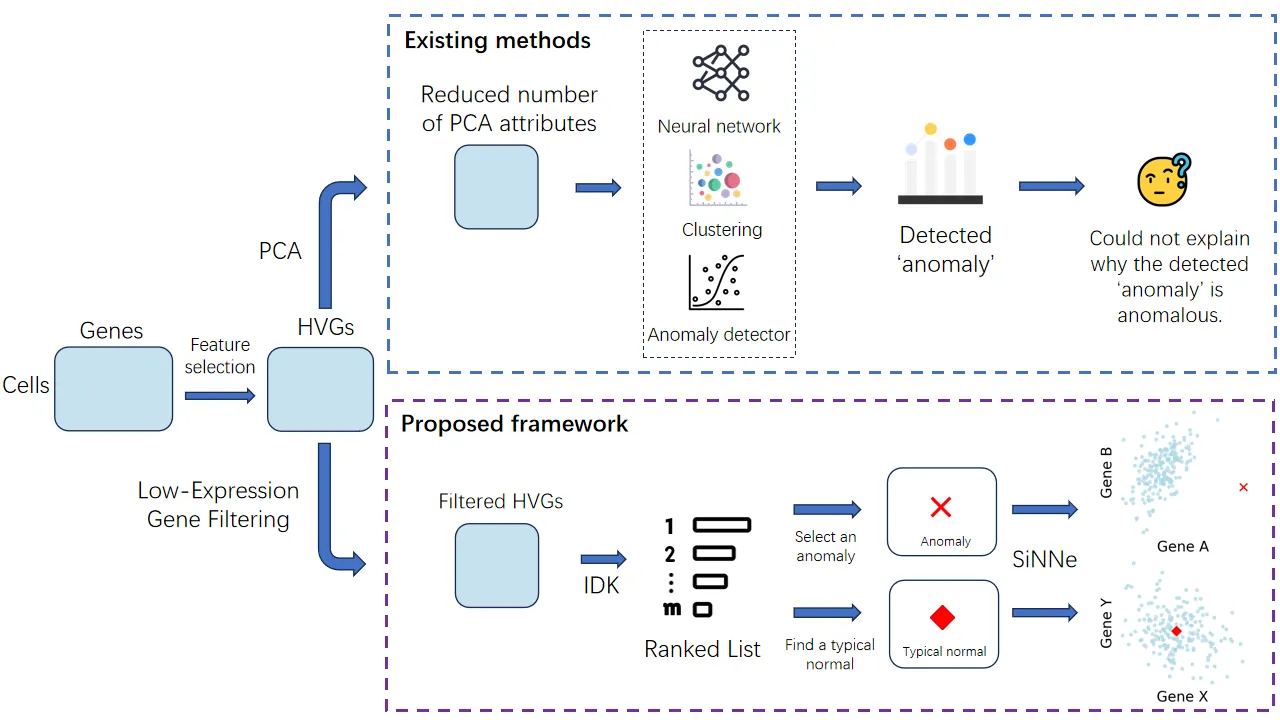
The detection of rare cell types in single-cell transcriptomics data is crucial for elucidating disease pathogenesis and tissue development dynamics. However, a critical gap that persists in current methods is their inability to provide an explanation based on genes for each cell they have detected as rare. We identify three primary sources of this deficiency. First, the anomaly detectors often function as "black boxes", designed to detect anomalies but unable to explain why a cell is anomalous. Second, the standard analytical framework hinders interpretability by relying on dimensionality reduction techniques, such as Principal Component Analysis (PCA), which transform meaningful gene expression data into abstract, uninterpretable features. Finally, existing explanation algorithms cannot be readily applied to this domain, as single-cell data is characterized by high dimensionality, noise, and substantial sparsity. To overcome these limitations, we introduce a framework for explainable anomaly detection in single-cell transcriptomics data which not only identifies individual anomalies, but also provides a visual explanation based on genes that makes an instance anomalous. This framework has two key ingredients that are not existed in current methods applied in this domain. First, it eliminates the PCA step which is deemed to be an essential component in previous studies. Second, it employs the state-of-art anomaly detector and explainer as the efficient and effective means to find each rare cell and the relevant gene subspace in order to provide explanations for each rare cell as well as the typical normal cell associated with the rare cell's closest normal cells.
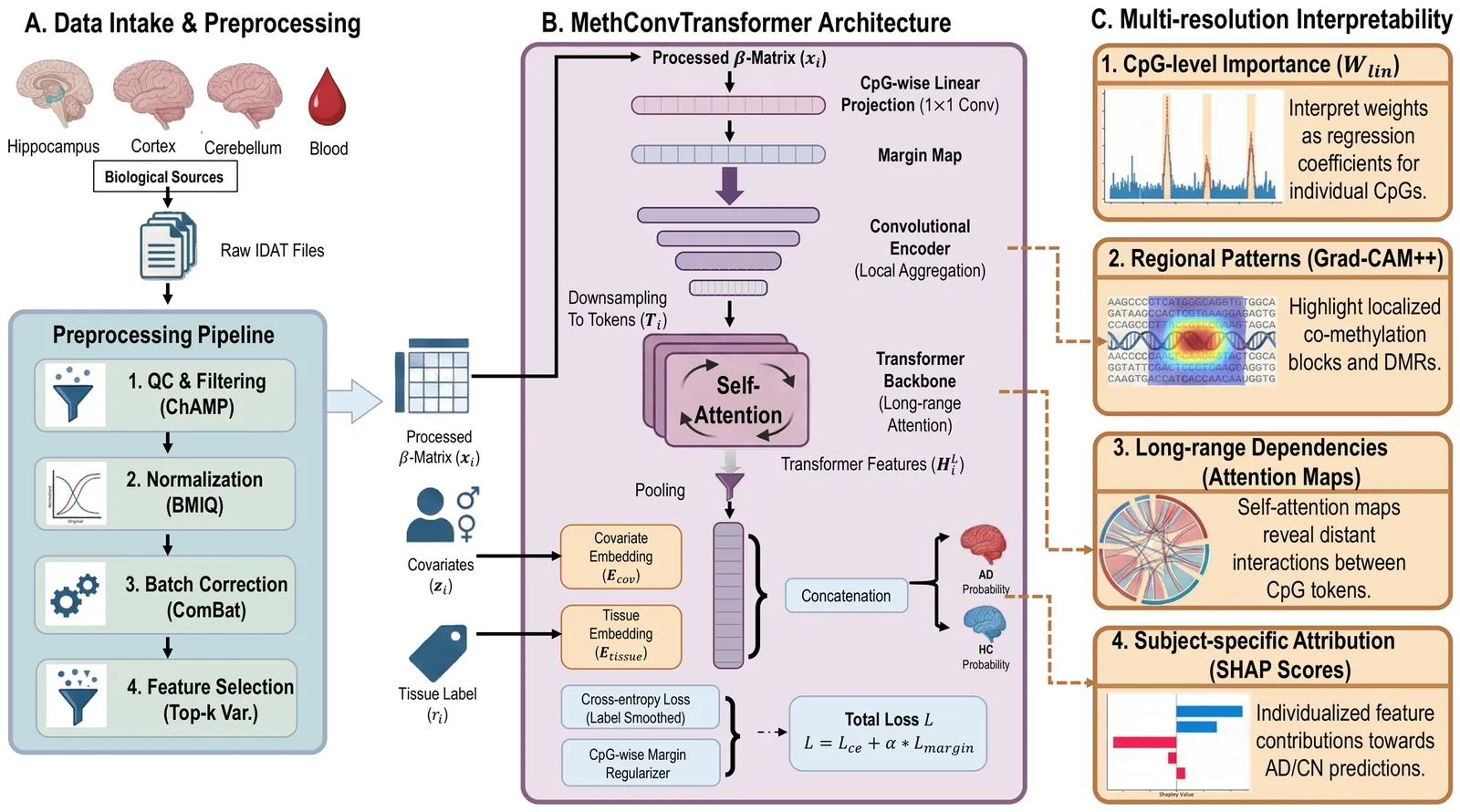
Alzheimer's disease (AD) is a multifactorial neurodegenerative disorder characterized by progressive cognitive decline and widespread epigenetic dysregulation in the brain. DNA methylation, as a stable yet dynamic epigenetic modification, holds promise as a noninvasive biomarker for early AD detection. However, methylation signatures vary substantially across tissues and studies, limiting reproducibility and translational utility. To address these challenges, we develop MethConvTransformer, a transformer-based deep learning framework that integrates DNA methylation profiles from both brain and peripheral tissues to enable biomarker discovery. The model couples a CpG-wise linear projection with convolutional and self-attention layers to capture local and long-range dependencies among CpG sites, while incorporating subject-level covariates and tissue embeddings to disentangle shared and region-specific methylation effects. In experiments across six GEO datasets and an independent ADNI validation cohort, our model consistently outperforms conventional machine-learning baselines, achieving superior discrimination and generalization. Moreover, interpretability analyses using linear projection, SHAP, and Grad-CAM++ reveal biologically meaningful methylation patterns aligned with AD-associated pathways, including immune receptor signaling, glycosylation, lipid metabolism, and endomembrane (ER/Golgi) organization. Together, these results indicate that MethConvTransformer delivers robust, cross-tissue epigenetic biomarkers for AD while providing multi-resolution interpretability, thereby advancing reproducible methylation-based diagnostics and offering testable hypotheses on disease mechanisms.
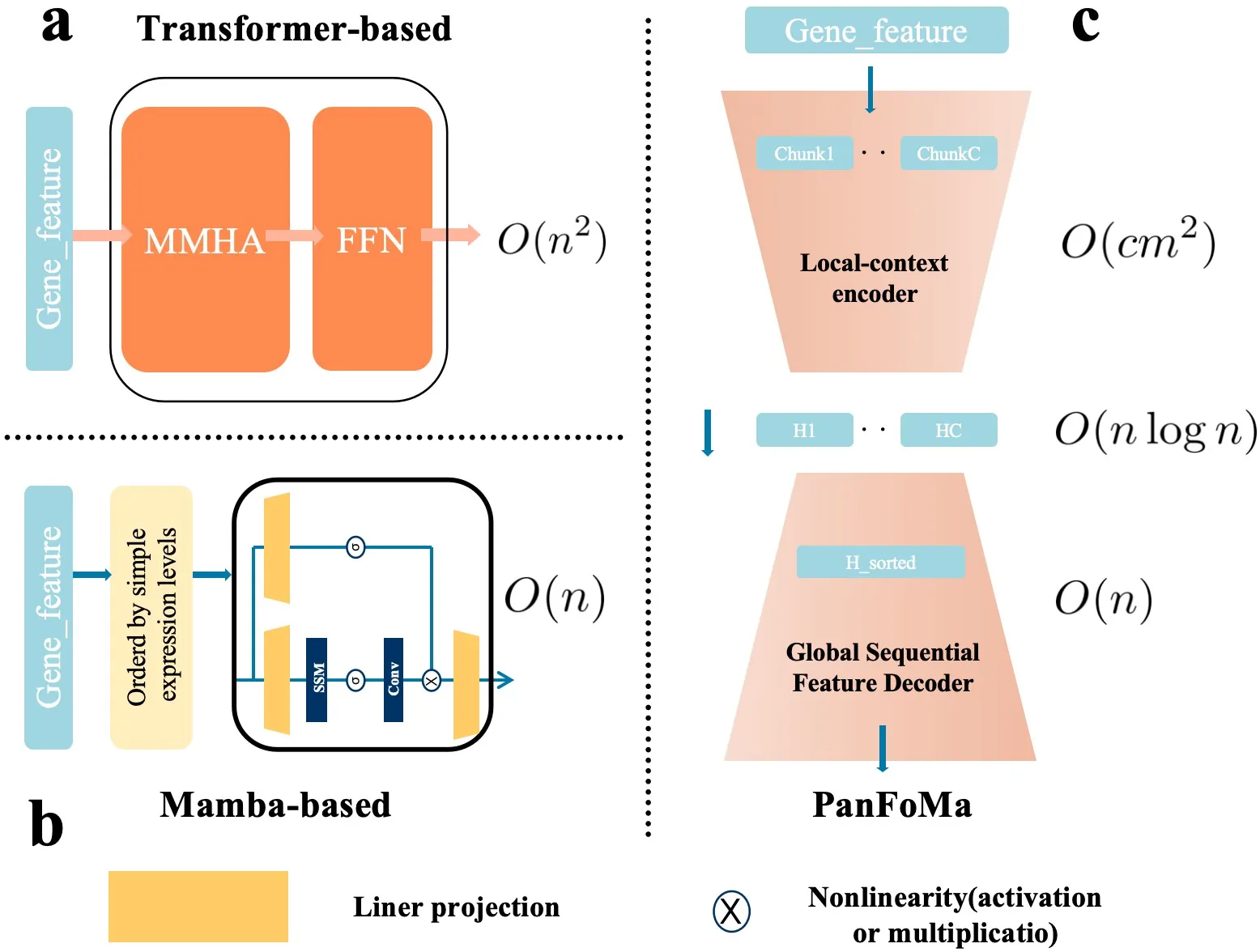
Single-cell RNA sequencing (scRNA-seq) is essential for decoding tumor heterogeneity. However, pan-cancer research still faces two key challenges: learning discriminative and efficient single-cell representations, and establishing a comprehensive evaluation benchmark. In this paper, we introduce PanFoMa, a lightweight hybrid neural network that combines the strengths of Transformers and state-space models to achieve a balance between performance and efficiency. PanFoMa consists of a front-end local-context encoder with shared self-attention layers to capture complex, order-independent gene interactions; and a back-end global sequential feature decoder that efficiently integrates global context using a linear-time state-space model. This modular design preserves the expressive power of Transformers while leveraging the scalability of Mamba to enable transcriptome modeling, effectively capturing both local and global regulatory signals. To enable robust evaluation, we also construct a large-scale pan-cancer single-cell benchmark, PanFoMaBench, containing over 3.5 million high-quality cells across 33 cancer subtypes, curated through a rigorous preprocessing pipeline. Experimental results show that PanFoMa outperforms state-of-the-art models on our pan-cancer benchmark (+4.0\%) and across multiple public tasks, including cell type annotation (+7.4\%), batch integration (+4.0\%) and multi-omics integration (+3.1\%). The code is available at https://github.com/Xiaoshui-Huang/PanFoMa.
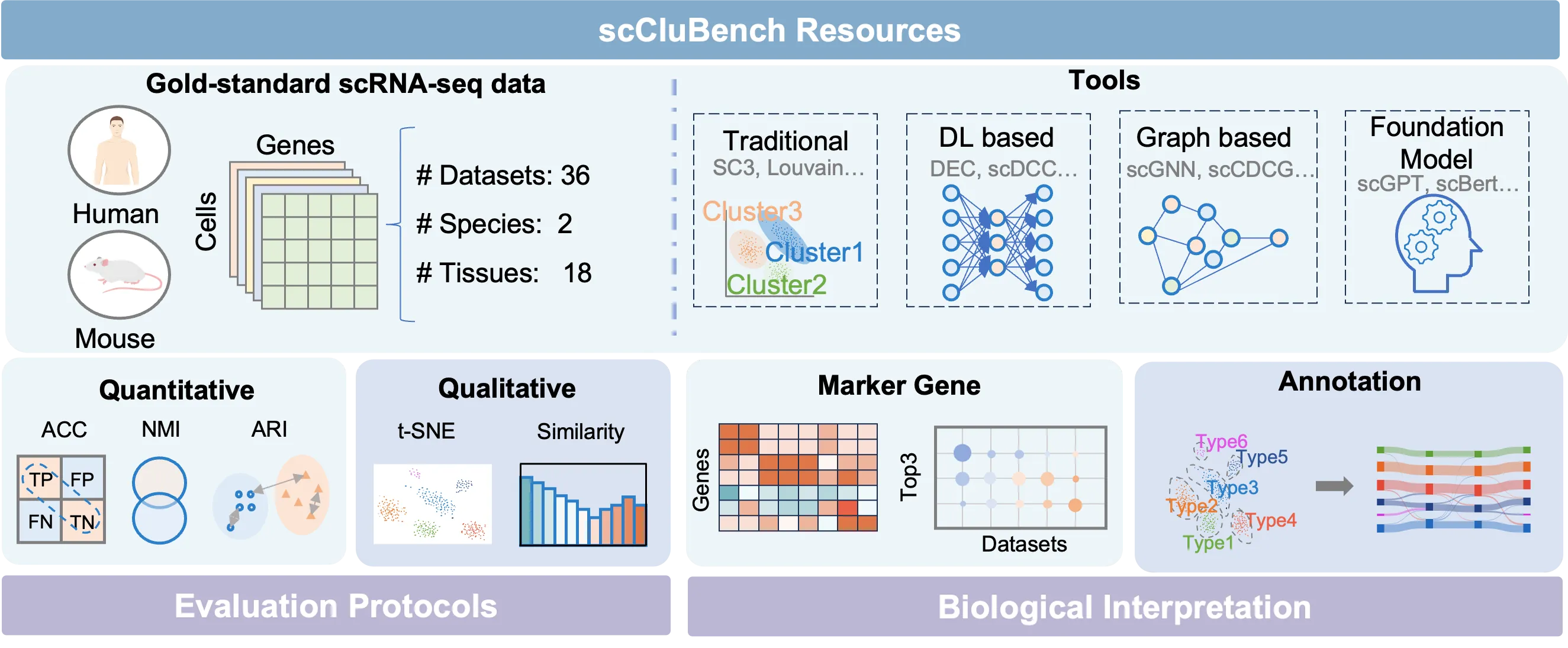
Cell clustering is crucial for uncovering cellular heterogeneity in single-cell RNA sequencing (scRNA-seq) data by identifying cell types and marker genes. Despite its importance, benchmarks for scRNA-seq clustering methods remain fragmented, often lacking standardized protocols and failing to incorporate recent advances in artificial intelligence. To fill these gaps, we present scCluBench, a comprehensive benchmark of clustering algorithms for scRNA-seq data. First, scCluBench provides 36 scRNA-seq datasets collected from diverse public sources, covering multiple tissues, which are uniformly processed and standardized to ensure consistency for systematic evaluation and downstream analyses. To evaluate performance, we collect and reproduce a range of scRNA-seq clustering methods, including traditional, deep learning-based, graph-based, and biological foundation models. We comprehensively evaluate each method both quantitatively and qualitatively, using core performance metrics as well as visualization analyses. Furthermore, we construct representative downstream biological tasks, such as marker gene identification and cell type annotation, to further assess the practical utility. scCluBench then investigates the performance differences and applicability boundaries of various clustering models across diverse analytical tasks, systematically assessing their robustness and scalability in real-world scenarios. Overall, scCluBench offers a standardized and user-friendly benchmark for scRNA-seq clustering, with curated datasets, unified evaluation protocols, and transparent analyses, facilitating informed method selection and providing valuable insights into model generalizability and application scope.
Modern genomic analyses increasingly rely on pangenomes, that is, representations of the genome of entire populations. The simplest representation of a pangenome is a set of individual genome sequences. Compared to e.g. sequence graphs, this has the advantage that efficient exact search via indexes based on the Burrows-Wheeler Transform (BWT) is possible, that no chimeric sequences are created, and that the results are not influenced by heuristics. However, such an index may report a match in thousands of positions even if these all correspond to the same locus, making downstream analysis unnecessarily expensive. For sufficiently similar sequences (e.g. human chromosomes), a multiple sequence alignment (MSA) can be computed. Since an MSA tends to group similar strings in the same columns, it is likely that a string occurring thousands of times in the pangenome can be described by very few columns in the MSA. We describe a method to tag entries in the BWT with the corresponding column in the MSA and develop an index that can map matches in the BWT to columns in the MSA in time proportional to the output. As a by-product, we can efficiently project a match to a designated reference genome, a capability that current pangenome aligners based on the BWT lack.
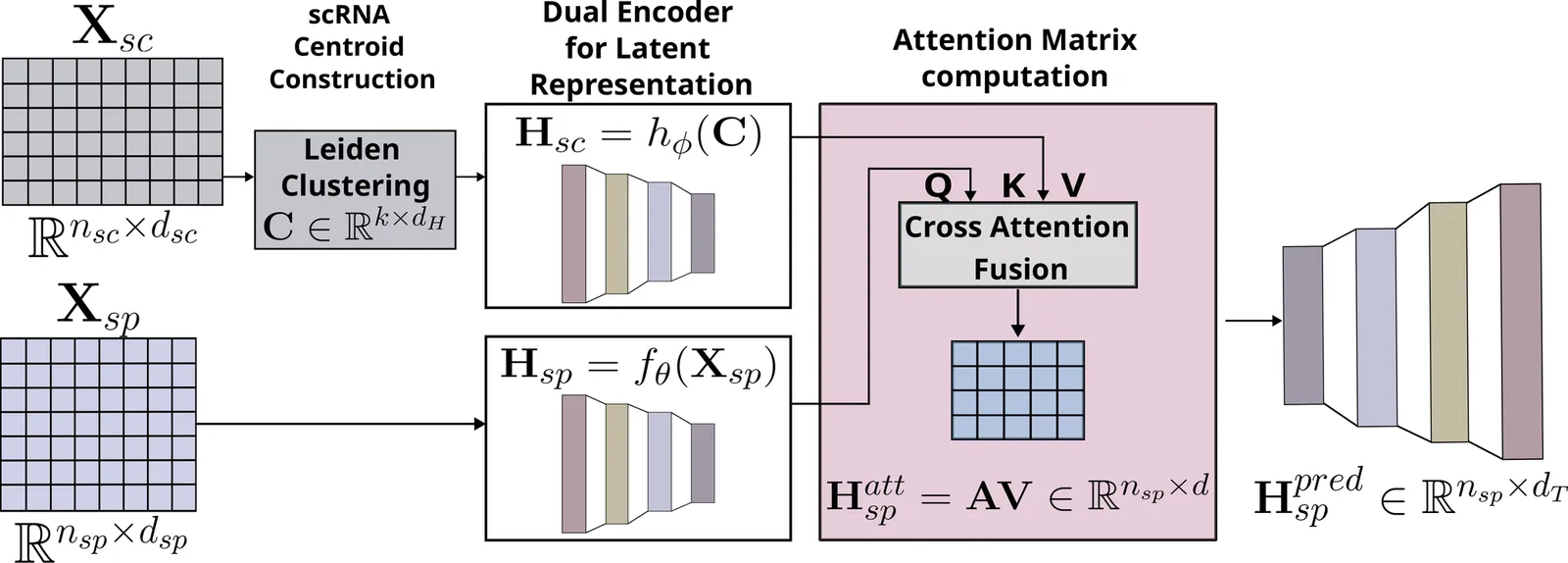
Spatial Transcriptomics enables mapping of gene expression within its native tissue context, but current platforms measure only a limited set of genes due to experimental constraints and excessive costs. To overcome this, computational models integrate Single-Cell RNA Sequencing data with Spatial Transcriptomics to predict unmeasured genes. We propose CASPER, a cross-attention based framework that predicts unmeasured gene expression in Spatial Transcriptomics by leveraging centroid-level representations from Single-Cell RNA Sequencing. We performed rigorous testing over four state-of-the-art Spatial Transcriptomics/Single-Cell RNA Sequencing dataset pairs across four existing baseline models. CASPER shows significant improvement in nine out of the twelve metrics for our experiments. This work paves the way for further work in Spatial Transcriptomics to Single-Cell RNA Sequencing modality translation. The code for CASPER is available at https://github.com/AI4Med-Lab/CASPER.
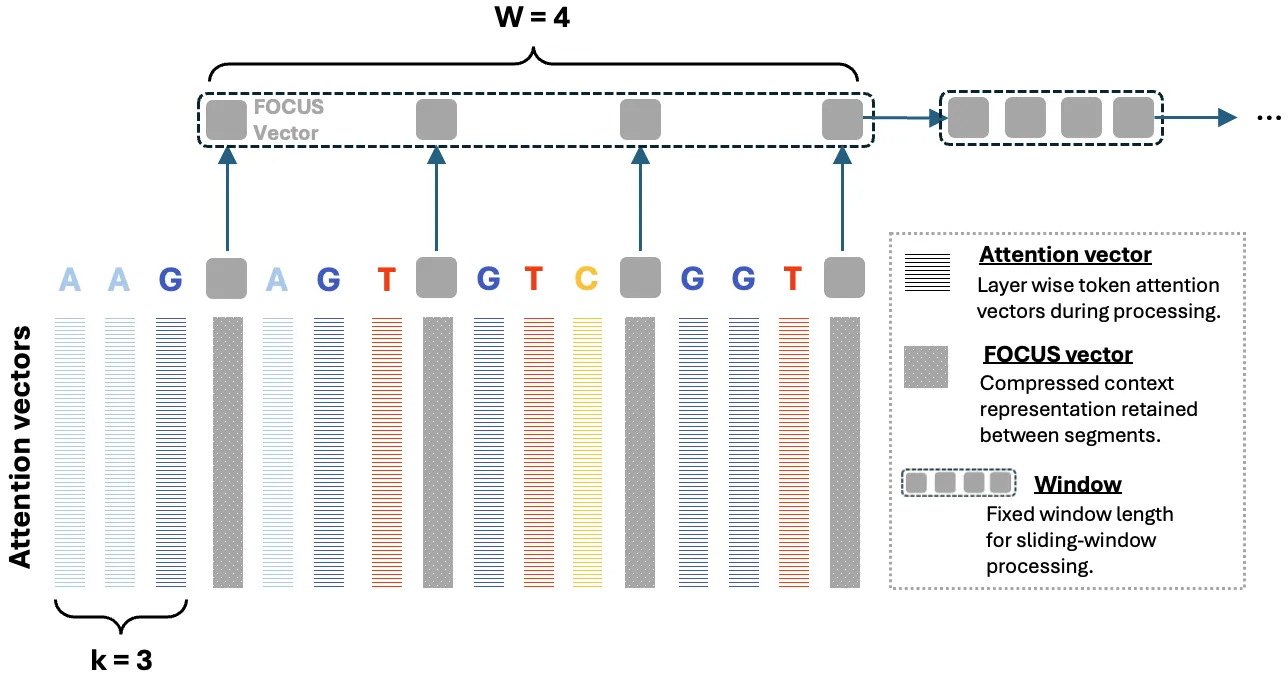
Trained on massive cross-species DNA corpora, DNA large language models (LLMs) learn the fundamental "grammar" and evolutionary patterns of genomic sequences. This makes them powerful priors for DNA sequence modeling, particularly over long ranges. However, two major constraints hinder their use in practice: the quadratic computational cost of self-attention and the growing memory required for key-value (KV) caches during autoregressive decoding. These constraints force the use of heuristics such as fixed-window truncation or sliding windows, which compromise fidelity on ultra-long sequences by discarding distant information. We introduce FOCUS (Feature-Oriented Compression for Ultra-long Self-attention), a progressive context-compression module that can be plugged into pretrained DNA LLMs. FOCUS combines the established k-mer representation in genomics with learnable hierarchical compression: it inserts summary tokens at k-mer granularity and progressively compresses attention key and value activations across multiple Transformer layers, retaining only the summary KV states across windows while discarding ordinary-token KV. A shared-boundary windowing scheme yields a stationary cross-window interface that propagates long-range information with minimal loss. We validate FOCUS on an Evo-2-based DNA LLM fine-tuned on GRCh38 chromosome 1 with self-supervised training and randomized compression schedules to promote robustness across compression ratios. On held-out human chromosomes, FOCUS achieves near-lossless fidelity: compressing a 1 kb context into only 10 summary tokens (about 100x) shifts the average per-nucleotide probability by only about 0.0004. Compared to a baseline without compression, FOCUS reduces KV-cache memory and converts effective inference scaling from O(N^2) to near-linear O(N), enabling about 100x longer inference windows on commodity GPUs with near-lossless fidelity.
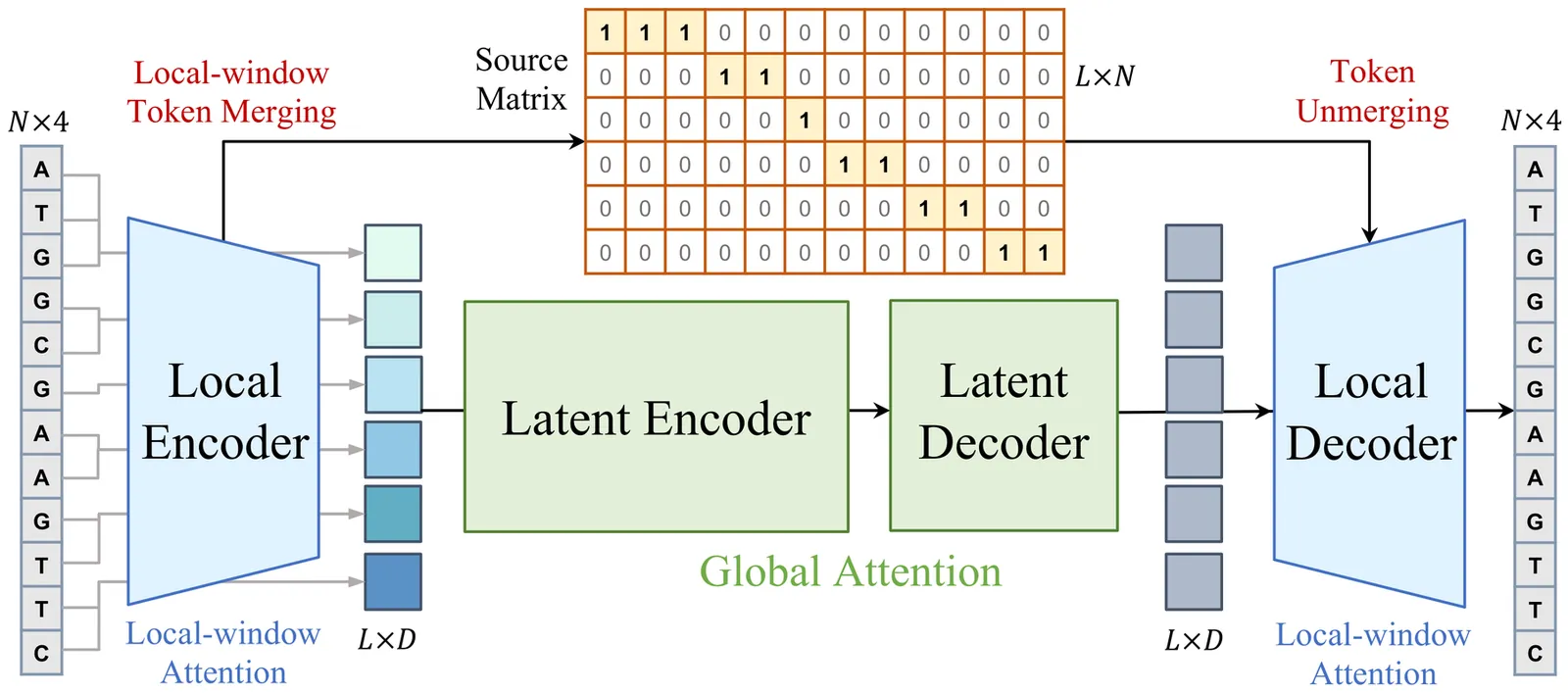
Modeling genomic sequences faces two unsolved challenges: the information density varies widely across different regions, while there is no clearly defined minimum vocabulary unit. Relying on either four primitive bases or independently designed DNA tokenizers, existing approaches with naive masked language modeling pre-training often fail to adapt to the varying complexities of genomic sequences. Leveraging Token Merging techniques, this paper introduces a hierarchical architecture that jointly optimizes a dynamic genomic tokenizer and latent Transformers with context-aware pre-training tasks. As for network structures, the tokenization module automatically chunks adjacent bases into words by stacking multiple layers of the differentiable token merging blocks with local-window constraints, then a Latent Encoder captures the global context of these merged words by full-attention blocks. Symmetrically employing a Latent Decoder and a Local Decoder, MergeDNA learns with two pre-training tasks: Merged Token Reconstruction simultaneously trains the dynamic tokenization module and adaptively filters important tokens, while Adaptive Masked Token Modeling learns to predict these filtered tokens to capture informative contents. Extensive experiments show that MergeDNA achieves superior performance on three popular DNA benchmarks and several multi-omics tasks with fine-tuning or zero-shot evaluation, outperforming typical tokenization methods and large-scale DNA foundation models.
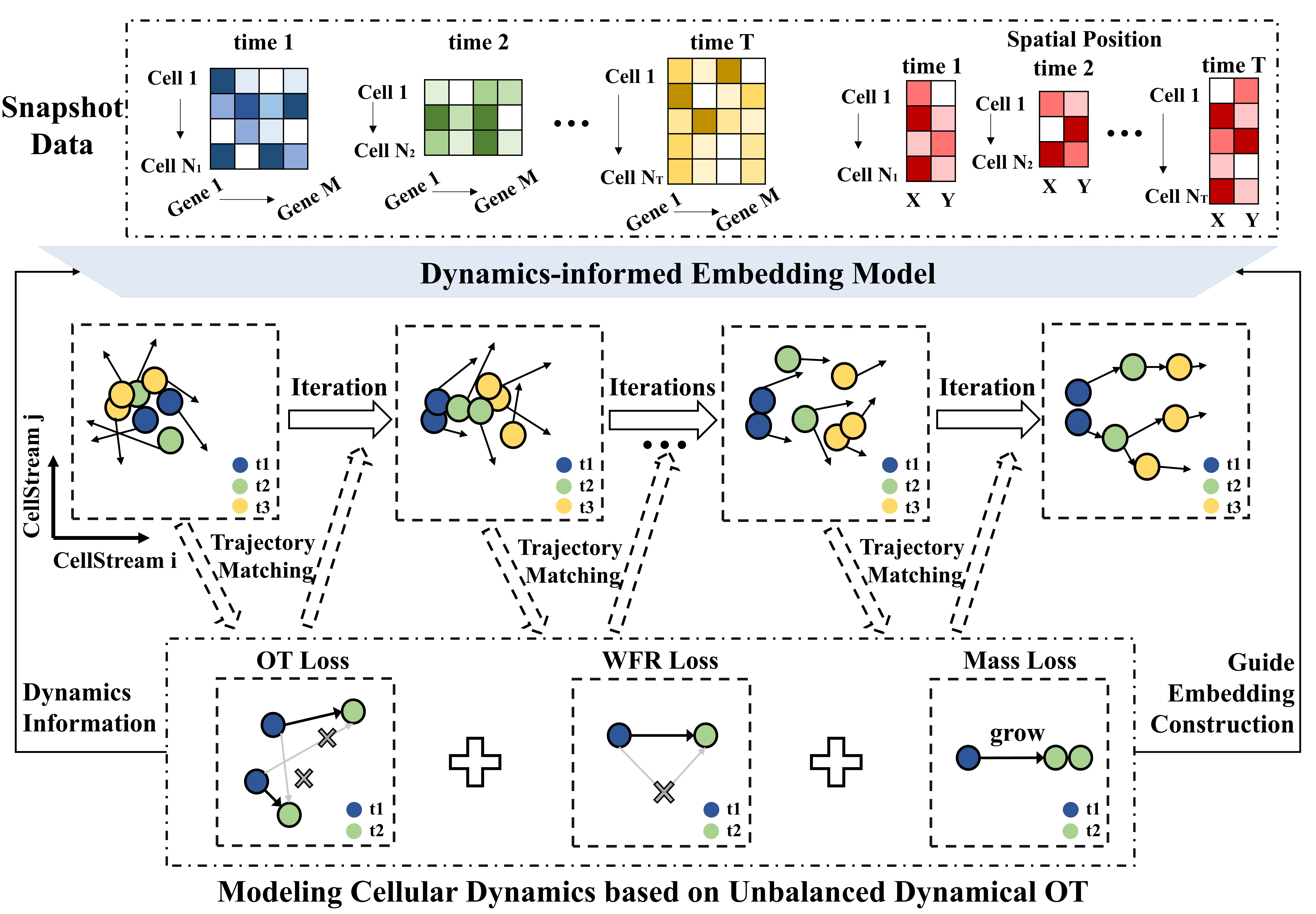
Single-cell RNA sequencing (scRNA-seq), especially temporally resolved datasets, enables genome-wide profiling of gene expression dynamics at single-cell resolution across discrete time points. However, current technologies provide only sparse, static snapshots of cell states and are inherently influenced by technical noise, complicating the inference and representation of continuous transcriptional dynamics. Although embedding methods can reduce dimensionality and mitigate technical noise, the majority of existing approaches typically treat trajectory inference separately from embedding construction, often neglecting temporal structure. To address this challenge, here we introduce CellStream, a novel deep learning framework that jointly learns embedding and cellular dynamics from single-cell snapshot data by integrating an autoencoder with unbalanced dynamical optimal transport. Compared to existing methods, CellStream generates dynamics-informed embeddings that robustly capture temporal developmental processes while maintaining high consistency with the underlying data manifold. We demonstrate CellStream's effectiveness on both simulated datasets and real scRNA-seq data, including spatial transcriptomics. Our experiments indicate significant quantitative improvements over state-of-the-art methods in representing cellular trajectories with enhanced temporal coherence and reduced noise sensitivity. Overall, CellStream provides a new tool for learning and representing continuous streams from the noisy, static snapshots of single-cell gene expression.
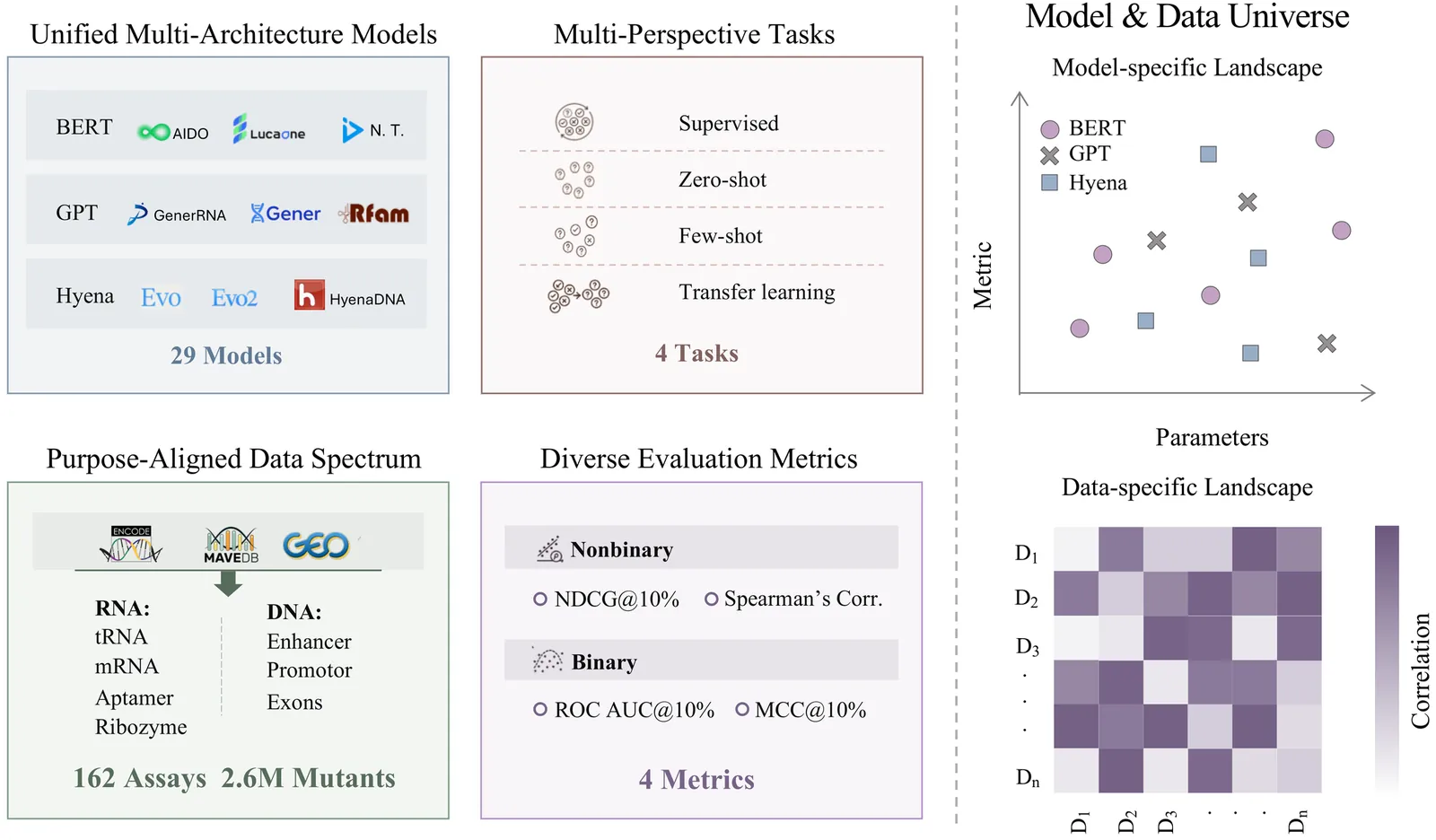
Nucleotide sequence variation can induce significant shifts in functional fitness. Recent nucleotide foundation models promise to predict such fitness effects directly from sequence, yet heterogeneous datasets and inconsistent preprocessing make it difficult to compare methods fairly across DNA and RNA families. Here we introduce NABench, a large-scale, systematic benchmark for nucleic acid fitness prediction. NABench aggregates 162 high-throughput assays and curates 2.6 million mutated sequences spanning diverse DNA and RNA families, with standardized splits and rich metadata. We show that NABench surpasses prior nucleotide fitness benchmarks in scale, diversity, and data quality. Under a unified evaluation suite, we rigorously assess 29 representative foundation models across zero-shot, few-shot prediction, transfer learning, and supervised settings. The results quantify performance heterogeneity across tasks and nucleic-acid types, demonstrating clear strengths and failure modes for different modeling choices and establishing strong, reproducible baselines. We release NABench to advance nucleic acid modeling, supporting downstream applications in RNA/DNA design, synthetic biology, and biochemistry. Our code is available at https://github.com/mrzzmrzz/NABench.
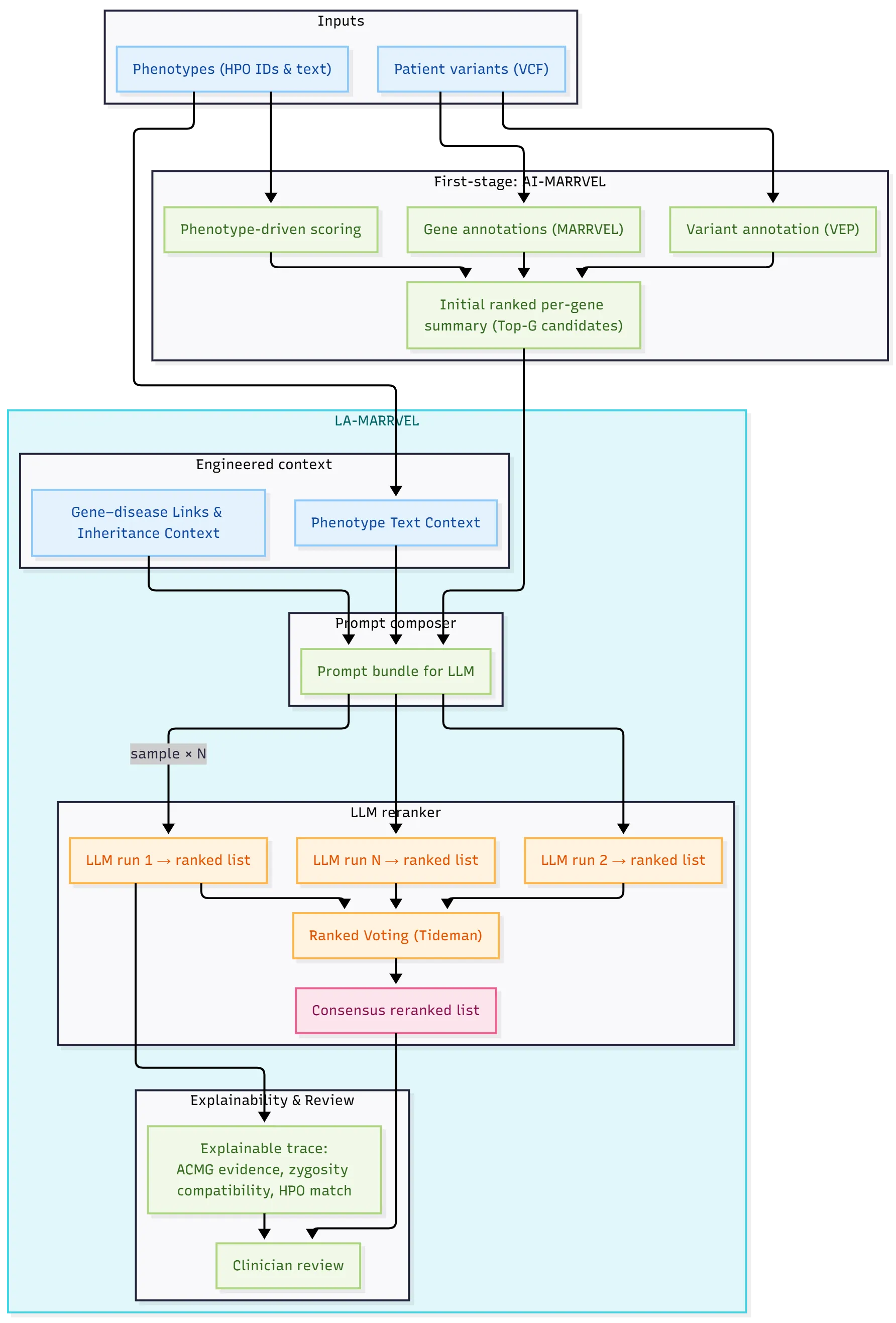
Diagnosing rare diseases requires linking gene findings with often unstructured reference text. Current pipelines collect many candidate genes, but clinicians still spend a lot of time filtering false positives and combining evidence from papers and databases. A key challenge is language: phenotype descriptions and inheritance patterns are written in prose, not fully captured by tables. Large language models (LLMs) can read such text, but clinical use needs grounding in citable knowledge and stable, repeatable behavior. We explore a knowledge-grounded and language-aware reranking layer on top of a high-recall first-stage pipeline. The goal is to improve precision and explainability, not to replace standard bioinformatics steps. We use expert-built context and a consensus method to reduce LLM variability, producing shorter, better-justified gene lists for expert review. LA-MARRVEL achieves the highest accuracy, outperforming other methods -- including traditional bioinformatics diagnostic tools (AI-MARRVEL, Exomiser, LIRICAL) and naive large language models (e.g., Anthropic Claude) -- with an average Recall@5 of 94.10%, a +3.65 percentage-point improvement over AI-MARRVEL. The LLM-generated reasoning provides clear prose on phenotype matching and inheritance patterns, making clinical review faster and easier. LA-MARRVEL has three parts: expert-engineered context that enriches phenotype and disease information; a ranked voting algorithm that combines multiple LLM runs to choose a consensus ranked gene list; and the AI-MARRVEL pipeline that provides first-stage ranks and gene annotations, already known as a state-of-the-art method in Rare Disease Diagnosis on BG, DDD, and UDN cohorts. The online AI-MARRVEL includes LA-MARRVEL as an LLM feature at https://ai.marrvel.org . We evaluate LA-MARRVEL on three datasets from independent cohorts of real-world diagnosed patients.

Introduction: Epigenomic datasets from high-throughput sequencing experiments are commonly summarized as genomic intervals. As the volume of this data grows, so does interest in analyzing it through deep learning. However, the heterogeneity of genomic interval data, where each dataset defines its own regions, creates barriers for machine learning methods that require consistent, discrete vocabularies. Methods: We introduce gtars-tokenizers, a high-performance library that maps genomic intervals to a predefined universe or vocabulary of regions, analogous to text tokenization in natural language processing. Built in Rust with bindings for Python, R, CLI, and WebAssembly, gtars-tokenizers implements two overlap methods (BITS and AIList) and integrates seamlessly with modern ML frameworks through Hugging Face-compatible APIs. Results: The gtars-tokenizers package achieves top efficiency for large-scale datasets, while enabling genomic intervals to be processed using standard ML workflows in PyTorch and TensorFlow without ad hoc preprocessing. This token-based approach bridges genomics and machine learning, supporting scalable and standardized analysis of interval data across diverse computational environments. Availability: PyPI and GitHub: https://github.com/databio/gtars.
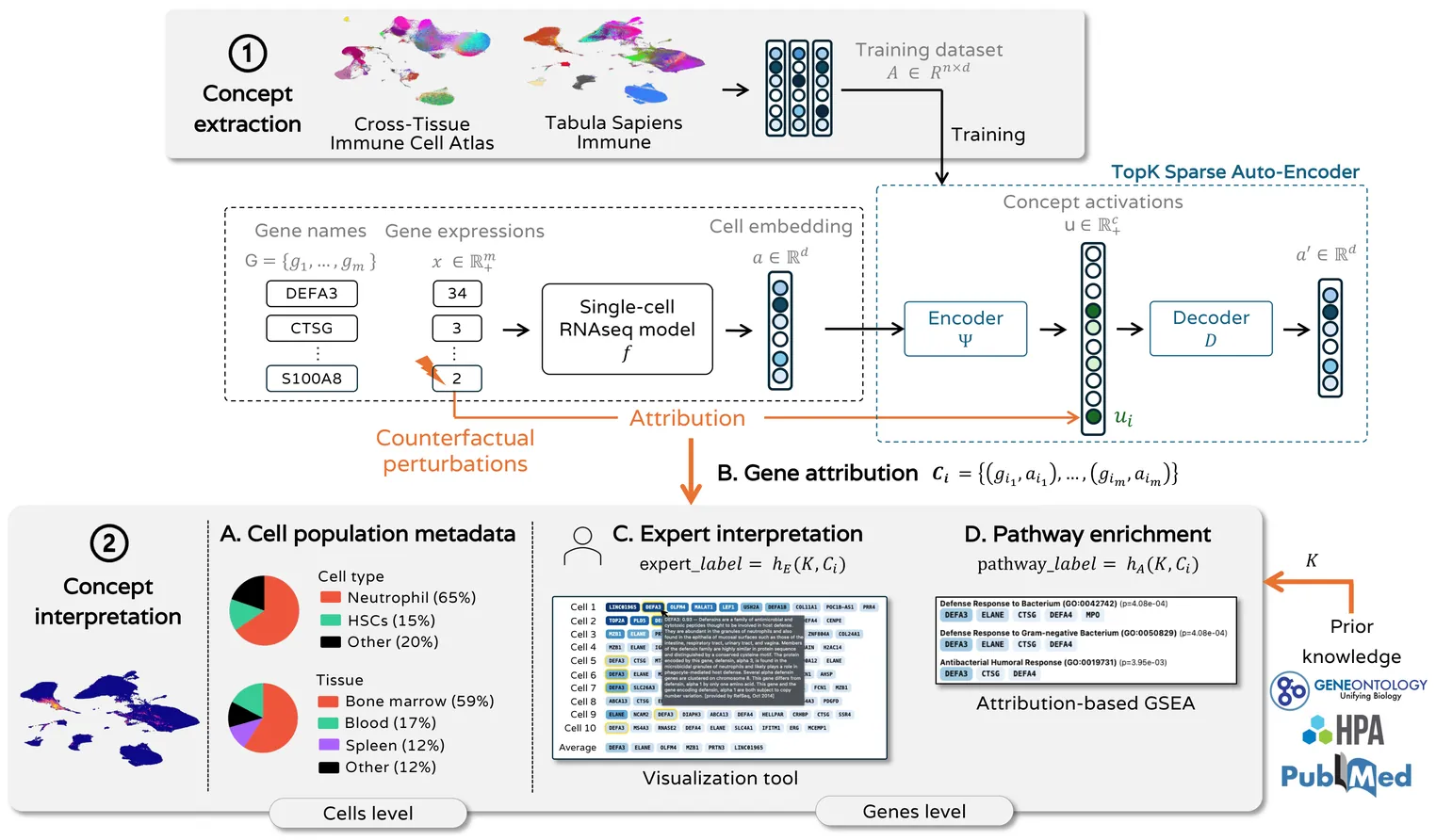
Single-cell RNA-seq foundation models achieve strong performance on downstream tasks but remain black boxes, limiting their utility for biological discovery. Recent work has shown that sparse dictionary learning can extract concepts from deep learning models, with promising applications in biomedical imaging and protein models. However, interpreting biological concepts remains challenging, as biological sequences are not inherently human-interpretable. We introduce a novel concept-based interpretability framework for single-cell RNA-seq models with a focus on concept interpretation and evaluation. We propose an attribution method with counterfactual perturbations that identifies genes that influence concept activation, moving beyond correlational approaches like differential expression analysis. We then provide two complementary interpretation approaches: an expert-driven analysis facilitated by an interactive interface and an ontology-driven method with attribution-based biological pathway enrichment. Applying our framework to two well-known single-cell RNA-seq models from the literature, we interpret concepts extracted by Top-K Sparse Auto-Encoders trained on two immune cell datasets. With a domain expert in immunology, we show that concepts improve interpretability compared to individual neurons while preserving the richness and informativeness of the latent representations. This work provides a principled framework for interpreting what biological knowledge foundation models have encoded, paving the way for their use for hypothesis generation and discovery.
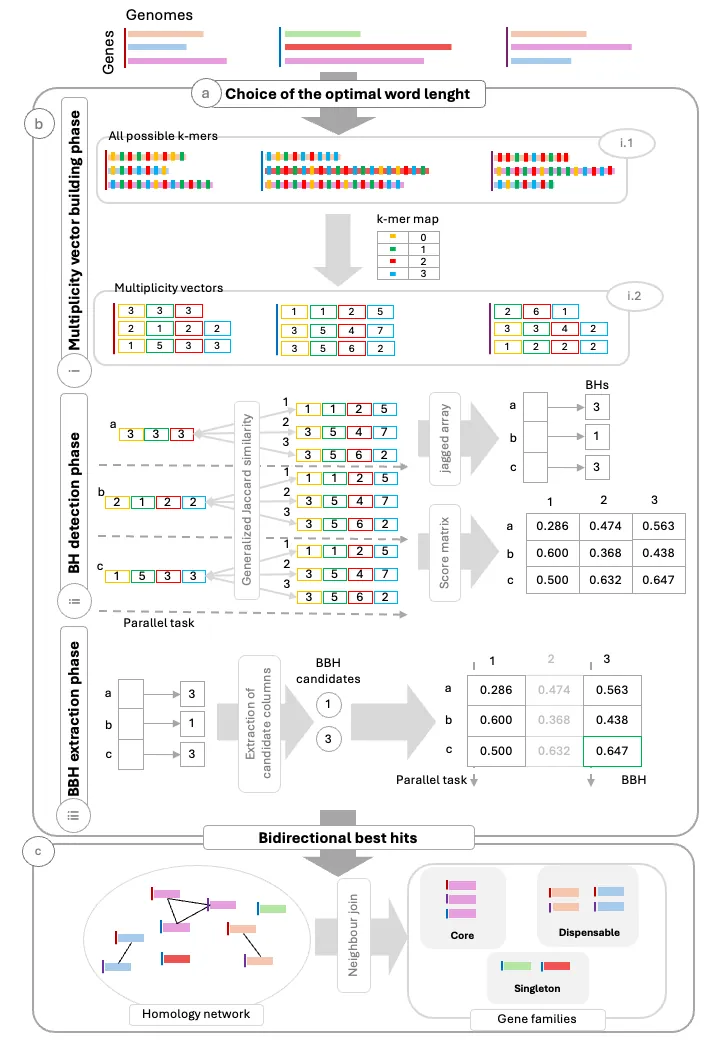
The identification of homologous gene families across multiple genomes is a central task in bacterial pangenomics traditionally requiring computationally demanding all-against-all comparisons. PanDelos addresses this challenge with an alignment-free and parameter-free approach based on k-mer profiles, combining high speed, ease of use, and competitive accuracy with state-of-the-art methods. However, the increasing availability of genomic data requires tools that can scale efficiently to larger datasets. To address this need, we present PanDelos-plus, a fully parallel, gene-centric redesign of PanDelos. The algorithm parallelizes the most computationally intensive phases (Best Hit detection and Bidirectional Best Hit extraction) through data decomposition and a thread pool strategy, while employing lightweight data structures to reduce memory usage. Benchmarks on synthetic datasets show that PanDelos-plus achieves up to 14x faster execution and reduces memory usage by up to 96%, while maintaining accuracy. These improvements enable population-scale comparative genomics to be performed on standard multicore workstations, making large-scale bacterial pangenome analysis accessible for routine use in everyday research.

Cancer exhibits diverse and complex phenotypes driven by multifaceted molecular interactions. Recent biomedical research has emphasized the comprehensive study of such diseases by integrating multi-omics datasets (genome, proteome, transcriptome, epigenome). This approach provides an efficient method for identifying genetic variants associated with cancer and offers a deeper understanding of how the disease develops and spreads. However, it is challenging to comprehend complex interactions among the features of multi-omics datasets compared to single omics. In this paper, we analyze lung cancer multi-omics datasets from The Cancer Genome Atlas (TCGA). Using four statistical methods, LIMMA, the T test, Canonical Correlation Analysis (CCA), and the Wilcoxon test, we identified differentially expressed genes across gene expression, DNA methylation, and miRNA expression data. We then integrated these multi-omics data using the Kernel Machine Regression (KMR) approach. Our findings reveal significant interactions among the three omics: gene expression, miRNA expression, and DNA methylation in lung cancer. From our data analysis, we identified 38 genes significantly associated with lung cancer. From our data analysis, we identified 38 genes significantly associated with lung cancer. Among these, eight genes of highest ranking (PDGFRB, PDGFRA, SNAI1, ID1, FGF11, TNXB, ITGB1, ZIC1) were highlighted by rigorous statistical analysis. Furthermore, in silico studies identified three top-ranked potential candidate drugs (Selinexor, Orapred, and Capmatinib) that could play a crucial role in the treatment of lung cancer. These proposed drugs are also supported by the findings of other independent studies, which underscore their potential efficacy in the fight against lung cancer.
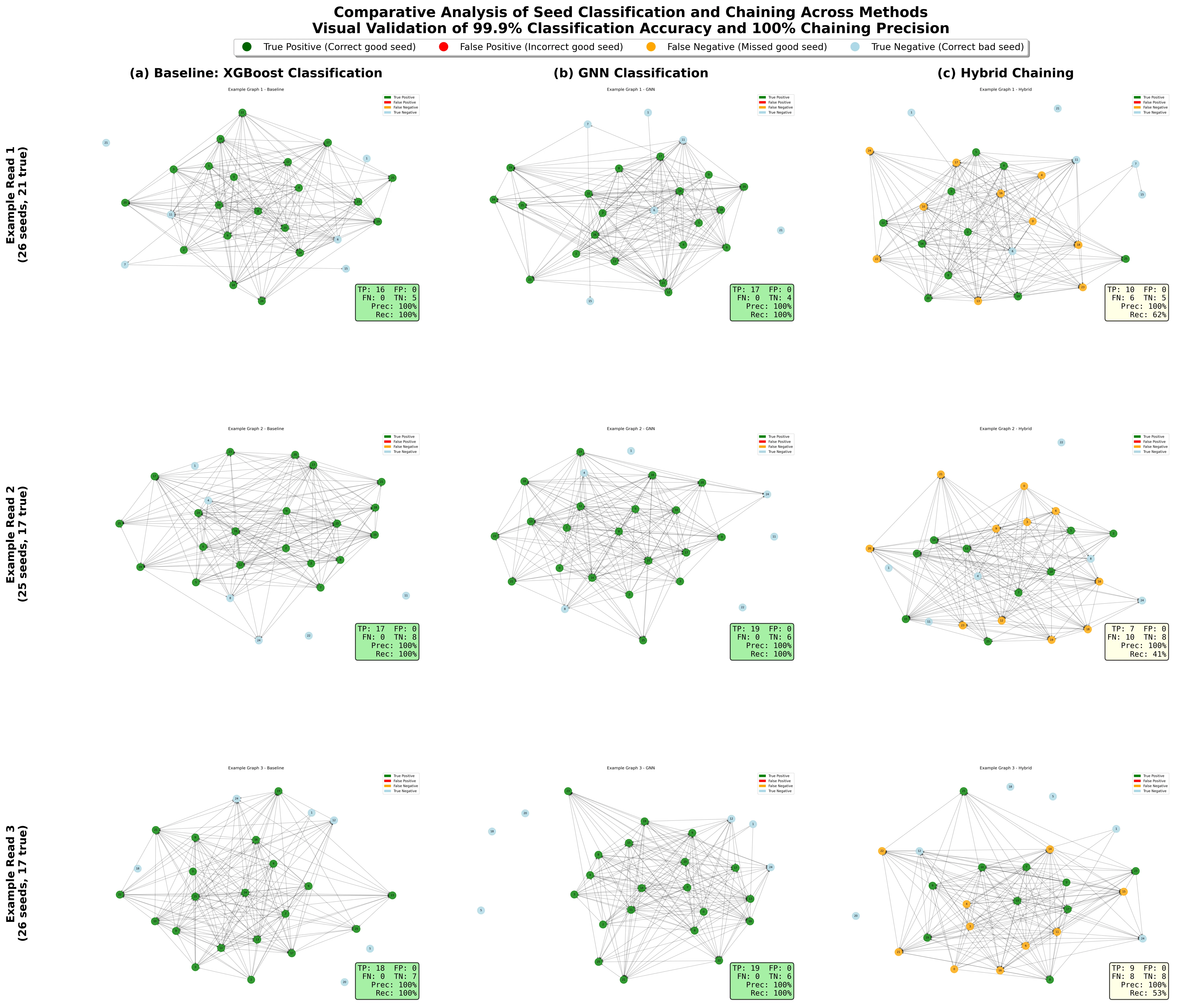
Nanopore sequencing enables real-time long-read DNA sequencing with reads exceeding 10 kilobases, but inherent error rates of 12-15 percent present significant computational challenges for read alignment. The critical seed chaining step must connect exact k-mer matches between reads and reference genomes while filtering spurious matches, yet state-of-the-art methods rely on fixed gap penalty functions unable to adapt to varying genomic contexts including tandem repeats and structural variants. This paper presents RawHash3, a hybrid framework combining graph neural networks with classical dynamic programming for adaptive seed chaining that maintains real-time performance while providing statistical guarantees. We formalize seed chaining as graph learning where seeds constitute nodes with 12-dimensional feature vectors and edges encode 8-dimensional spatial relationships including gap consistency. Our architecture employs three-layer EdgeConv GNN with confidence-based method selection that dynamically switches between learned guidance and algorithmic fallback. Comprehensive evaluation on 1,000 synthetic nanopore reads with 5,200 test seeds demonstrates RawHash3 achieves 99.94 percent precision and 40.07 percent recall, representing statistically significant 25.0 percent relative improvement over baseline with p less than 0.001. The system maintains median inference latency of 1.59ms meeting real-time constraints, while demonstrating superior robustness with 100 percent success rate under 20 percent label corruption versus baseline degradation to 30.3 percent. Cross-validation confirms stability establishing graph neural networks as viable approach for production genomics pipelines.
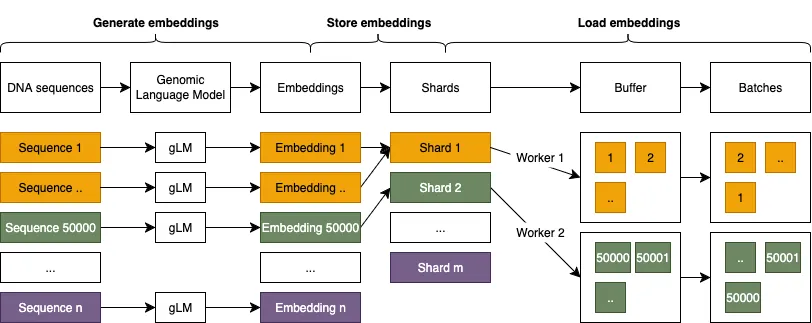
Large Language Models are increasingly popular in genomics due to their potential to decode complex biological sequences. Hence, researchers require a standardized benchmark to evaluate DNA Language Models (DNA LMs) capabilities. However, evaluating DNA LMs is a complex task that intersects genomic's domain-specific challenges and machine learning methodologies, where seemingly minor implementation details can significantly compromise benchmark validity. We demonstrate this through BEND (Benchmarking DNA Language Models), where hardware-dependent hyperparameters -- number of data loading workers and buffer sizes -- create spurious performance variations of up to 4% for identical models. The problem stems from inadequate data shuffling interacting with domain specific data characteristics. Experiments with three DNA language models (HyenaDNA, DNABERT-2, ResNet-LM) show these artifacts affect both absolute performance and relative model rankings. We propose a simple solution: pre-shuffling data before storage eliminates hardware dependencies while maintaining efficiency. This work highlights how standard ML practices can interact unexpectedly with domain-specific data characteristics, with broader implications for benchmark design in specialized domains.
Modern genomic analyses increasingly rely on pangenomes, that is, representations of the genome of entire populations. The simplest representation of a pangenome is a set of individual genome sequences. Compared to e.g. sequence graphs, this has the advantage that efficient exact search via indexes based on the Burrows-Wheeler Transform (BWT) is possible, that no chimeric sequences are created, and that the results are not influenced by heuristics. However, such an index may report a match in thousands of positions even if these all correspond to the same locus, making downstream analysis unnecessarily expensive. For sufficiently similar sequences (e.g. human chromosomes), a multiple sequence alignment (MSA) can be computed. Since an MSA tends to group similar strings in the same columns, it is likely that a string occurring thousands of times in the pangenome can be described by very few columns in the MSA. We describe a method to tag entries in the BWT with the corresponding column in the MSA and develop an index that can map matches in the BWT to columns in the MSA in time proportional to the output. As a by-product, we can efficiently project a match to a designated reference genome, a capability that current pangenome aligners based on the BWT lack.
Single-cell RNA sequencing (scRNA-seq), especially temporally resolved datasets, enables genome-wide profiling of gene expression dynamics at single-cell resolution across discrete time points. However, current technologies provide only sparse, static snapshots of cell states and are inherently influenced by technical noise, complicating the inference and representation of continuous transcriptional dynamics. Although embedding methods can reduce dimensionality and mitigate technical noise, the majority of existing approaches typically treat trajectory inference separately from embedding construction, often neglecting temporal structure. To address this challenge, here we introduce CellStream, a novel deep learning framework that jointly learns embedding and cellular dynamics from single-cell snapshot data by integrating an autoencoder with unbalanced dynamical optimal transport. Compared to existing methods, CellStream generates dynamics-informed embeddings that robustly capture temporal developmental processes while maintaining high consistency with the underlying data manifold. We demonstrate CellStream's effectiveness on both simulated datasets and real scRNA-seq data, including spatial transcriptomics. Our experiments indicate significant quantitative improvements over state-of-the-art methods in representing cellular trajectories with enhanced temporal coherence and reduced noise sensitivity. Overall, CellStream provides a new tool for learning and representing continuous streams from the noisy, static snapshots of single-cell gene expression.
Introduction: Epigenomic datasets from high-throughput sequencing experiments are commonly summarized as genomic intervals. As the volume of this data grows, so does interest in analyzing it through deep learning. However, the heterogeneity of genomic interval data, where each dataset defines its own regions, creates barriers for machine learning methods that require consistent, discrete vocabularies. Methods: We introduce gtars-tokenizers, a high-performance library that maps genomic intervals to a predefined universe or vocabulary of regions, analogous to text tokenization in natural language processing. Built in Rust with bindings for Python, R, CLI, and WebAssembly, gtars-tokenizers implements two overlap methods (BITS and AIList) and integrates seamlessly with modern ML frameworks through Hugging Face-compatible APIs. Results: The gtars-tokenizers package achieves top efficiency for large-scale datasets, while enabling genomic intervals to be processed using standard ML workflows in PyTorch and TensorFlow without ad hoc preprocessing. This token-based approach bridges genomics and machine learning, supporting scalable and standardized analysis of interval data across diverse computational environments. Availability: PyPI and GitHub: https://github.com/databio/gtars.
Cancer exhibits diverse and complex phenotypes driven by multifaceted molecular interactions. Recent biomedical research has emphasized the comprehensive study of such diseases by integrating multi-omics datasets (genome, proteome, transcriptome, epigenome). This approach provides an efficient method for identifying genetic variants associated with cancer and offers a deeper understanding of how the disease develops and spreads. However, it is challenging to comprehend complex interactions among the features of multi-omics datasets compared to single omics. In this paper, we analyze lung cancer multi-omics datasets from The Cancer Genome Atlas (TCGA). Using four statistical methods, LIMMA, the T test, Canonical Correlation Analysis (CCA), and the Wilcoxon test, we identified differentially expressed genes across gene expression, DNA methylation, and miRNA expression data. We then integrated these multi-omics data using the Kernel Machine Regression (KMR) approach. Our findings reveal significant interactions among the three omics: gene expression, miRNA expression, and DNA methylation in lung cancer. From our data analysis, we identified 38 genes significantly associated with lung cancer. From our data analysis, we identified 38 genes significantly associated with lung cancer. Among these, eight genes of highest ranking (PDGFRB, PDGFRA, SNAI1, ID1, FGF11, TNXB, ITGB1, ZIC1) were highlighted by rigorous statistical analysis. Furthermore, in silico studies identified three top-ranked potential candidate drugs (Selinexor, Orapred, and Capmatinib) that could play a crucial role in the treatment of lung cancer. These proposed drugs are also supported by the findings of other independent studies, which underscore their potential efficacy in the fight against lung cancer.
Nanopore sequencing enables real-time long-read DNA sequencing with reads exceeding 10 kilobases, but inherent error rates of 12-15 percent present significant computational challenges for read alignment. The critical seed chaining step must connect exact k-mer matches between reads and reference genomes while filtering spurious matches, yet state-of-the-art methods rely on fixed gap penalty functions unable to adapt to varying genomic contexts including tandem repeats and structural variants. This paper presents RawHash3, a hybrid framework combining graph neural networks with classical dynamic programming for adaptive seed chaining that maintains real-time performance while providing statistical guarantees. We formalize seed chaining as graph learning where seeds constitute nodes with 12-dimensional feature vectors and edges encode 8-dimensional spatial relationships including gap consistency. Our architecture employs three-layer EdgeConv GNN with confidence-based method selection that dynamically switches between learned guidance and algorithmic fallback. Comprehensive evaluation on 1,000 synthetic nanopore reads with 5,200 test seeds demonstrates RawHash3 achieves 99.94 percent precision and 40.07 percent recall, representing statistically significant 25.0 percent relative improvement over baseline with p less than 0.001. The system maintains median inference latency of 1.59ms meeting real-time constraints, while demonstrating superior robustness with 100 percent success rate under 20 percent label corruption versus baseline degradation to 30.3 percent. Cross-validation confirms stability establishing graph neural networks as viable approach for production genomics pipelines.
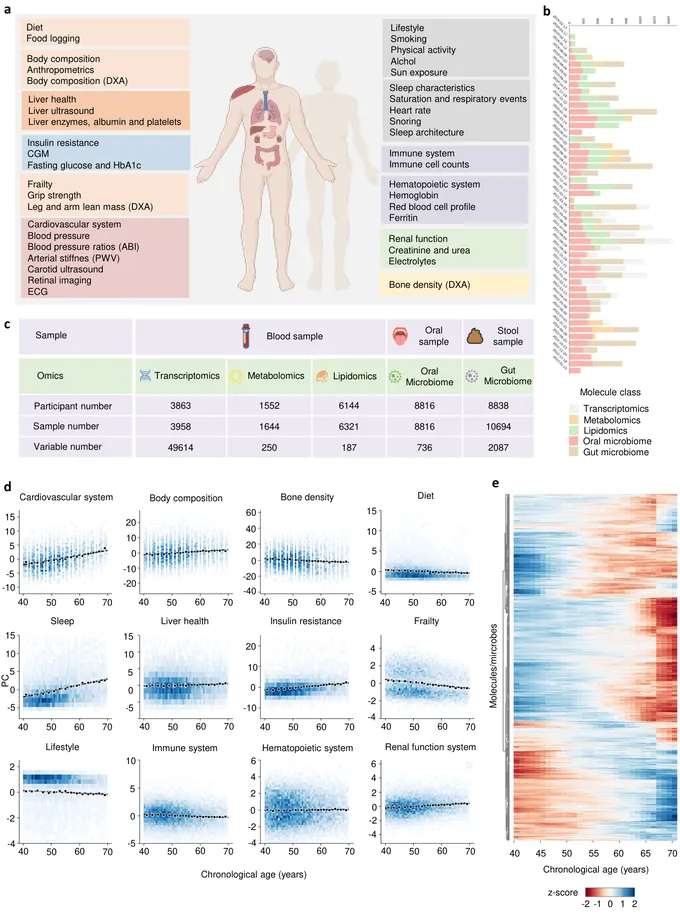
Aging is a highly complex and heterogeneous process that progresses at different rates across individuals, making biological age (BA) a more accurate indicator of physiological decline than chronological age. While previous studies have built aging clocks using single-omics data, they often fail to capture the full molecular complexity of human aging. In this work, we leveraged the Human Phenotype Project, a large-scale cohort of 10,000 adults aged 40-70 years, with extensive longitudinal profiling that includes clinical, behavioral, environmental, and multi-omics datasets spanning transcriptomics, lipidomics, metabolomics, and the microbiome. By employing advanced machine learning frameworks capable of modeling nonlinear biological dynamics, we developed and rigorously validated a multi-omics aging clock that robustly predicts diverse health outcomes and future disease risk. Unsupervised clustering of the integrated molecular profiles from multi-omics uncovered distinct biological subtypes of aging, revealing striking heterogeneity in aging trajectories and pinpointing pathway-specific alterations associated with different aging patterns. These findings demonstrate the power of multi-omics integration to decode the molecular landscape of aging and lay the groundwork for personalized healthspan monitoring and precision strategies to prevent age-related diseases.

Single-cell RNA sequencing (scRNA-seq) technology enables systematic delineation of cellular states and interactions, providing crucial insights into cellular heterogeneity. Building on this potential, numerous computational methods have been developed for tasks such as cell clustering, cell type annotation, and marker gene identification. To fully assess and compare these methods, standardized, analysis-ready datasets are essential. However, such datasets remain scarce, and variations in data formats, preprocessing workflows, and annotation strategies hinder reproducibility and complicate systematic evaluation of existing methods. To address these challenges, we present scUnified, an AI-ready standardized resource for single-cell RNA sequencing data that consolidates 13 high-quality datasets spanning two species (human and mouse) and nine tissue types. All datasets undergo standardized quality control and preprocessing and are stored in a uniform format to enable direct application in diverse computational analyses without additional data cleaning. We further demonstrate the utility of scUnified through experimental analyses of representative biological tasks, providing a reproducible foundation for the standardized evaluation of computational methods on a unified dataset.
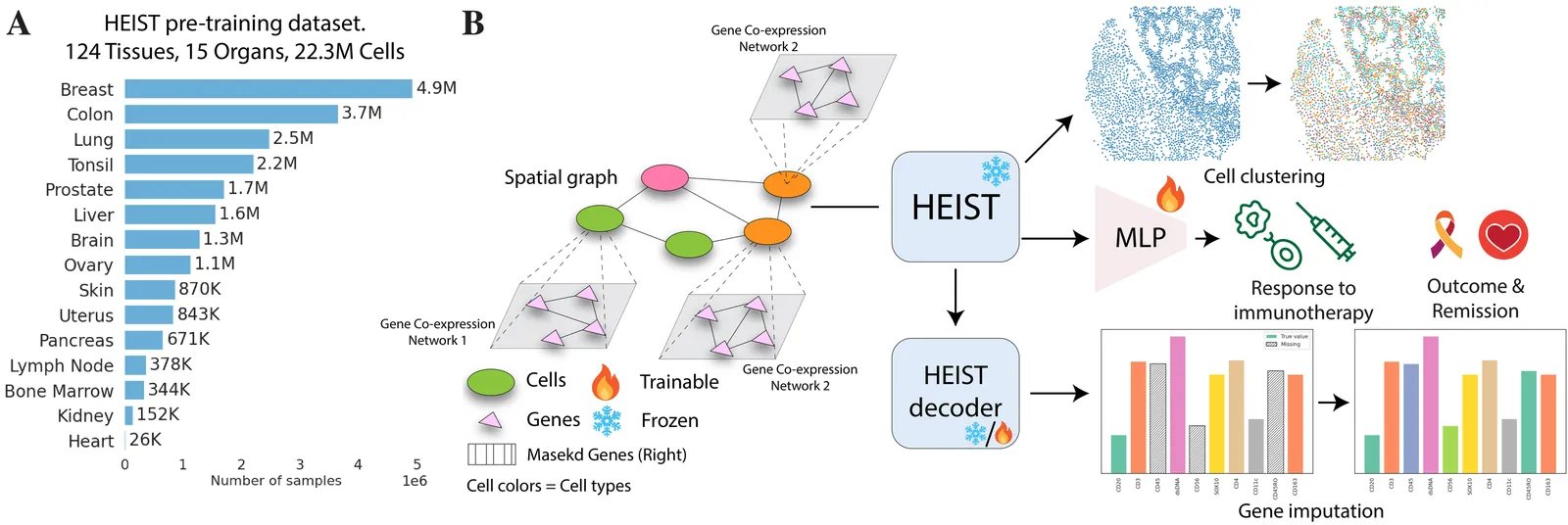
Single-cell transcriptomics and proteomics have become a great source for data-driven insights into biology, enabling the use of advanced deep learning methods to understand cellular heterogeneity and gene expression at the single-cell level. With the advent of spatial-omics data, we have the promise of characterizing cells within their tissue context as it provides both spatial coordinates and intra-cellular transcriptional or protein counts. Proteomics offers a complementary view by directly measuring proteins, which are the primary effectors of cellular function and key therapeutic targets. However, existing models either ignore the spatial information or the complex genetic and proteomic programs within cells. Thus they cannot infer how cell internal regulation adapts to microenvironmental cues. Furthermore, these models often utilize fixed gene vocabularies, hindering their generalizability unseen genes. In this paper, we introduce HEIST, a hierarchical graph transformer foundation model for spatial transcriptomics and proteomics. HEIST models tissues as hierarchical graphs. The higher level graph is a spatial cell graph, and each cell in turn, is represented by its lower level gene co-expression network graph. HEIST achieves this by performing both intra-level and cross-level message passing to utilize the hierarchy in its embeddings and can thus generalize to novel datatypes including spatial proteomics without retraining. HEIST is pretrained on 22.3M cells from 124 tissues across 15 organs using spatially-aware contrastive and masked autoencoding objectives. Unsupervised analysis of HEIST embeddings reveals spatially informed subpopulations missed by prior models. Downstream evaluations demonstrate generalizability to proteomics data and state-of-the-art performance in clinical outcome prediction, cell type annotation, and gene imputation across multiple technologies.
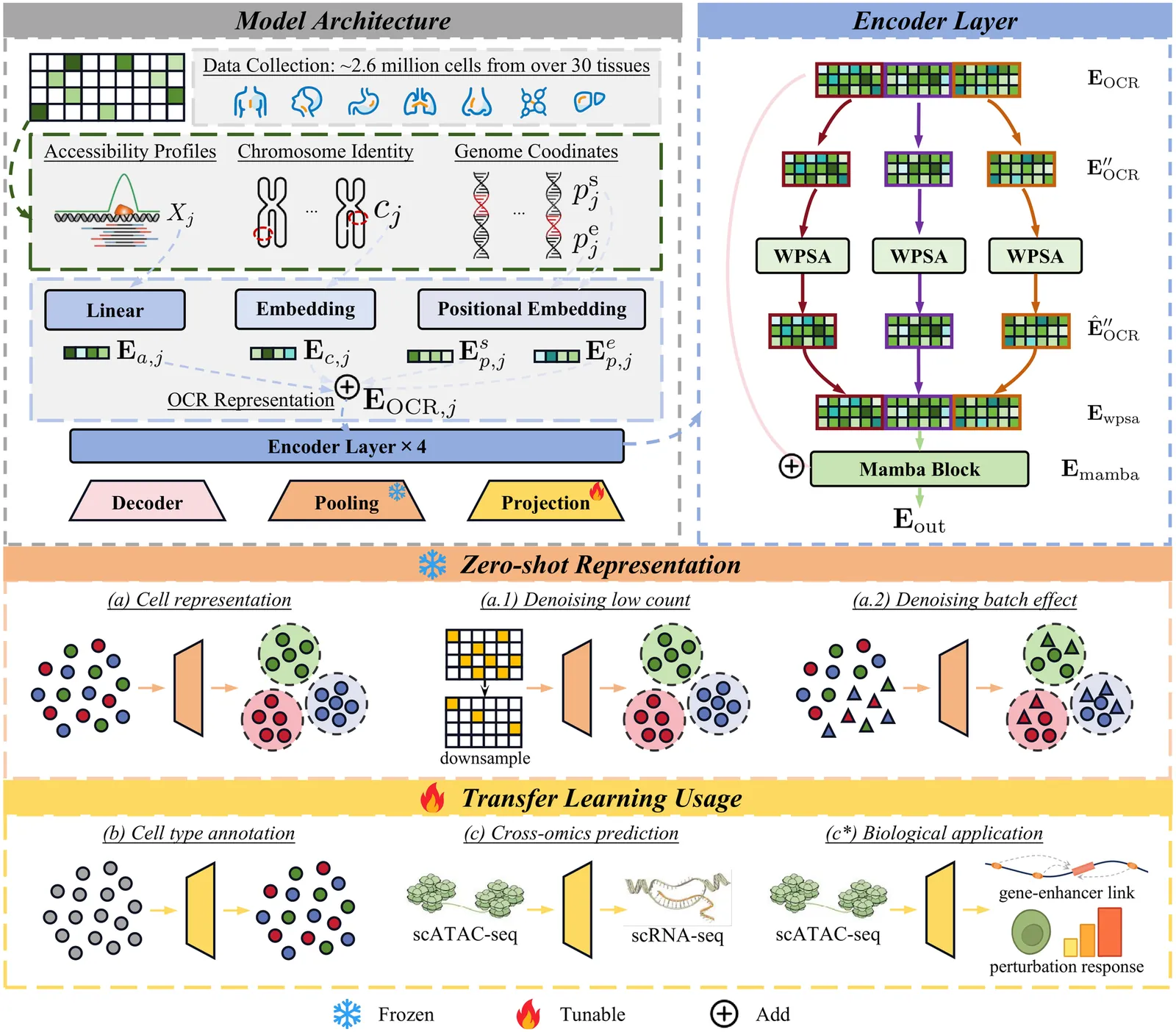
The advent of single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) offers an innovative perspective for deciphering regulatory mechanisms by assembling a vast repository of single-cell chromatin accessibility data. While foundation models have achieved significant success in single-cell transcriptomics, there is currently no foundation model for scATAC-seq that supports zero-shot high-quality cell identification and comprehensive multi-omics analysis simultaneously. Key challenges lie in the high dimensionality and sparsity of scATAC-seq data, as well as the lack of a standardized schema for representing open chromatin regions (OCRs). Here, we present ChromFound, a foundation model tailored for scATAC-seq. ChromFound utilizes a hybrid architecture and genome-aware tokenization to effectively capture genome-wide long contexts and regulatory signals from dynamic chromatin landscapes. Pretrained on 1.97 million cells from 30 tissues and 6 disease conditions, ChromFound demonstrates broad applicability across 6 diverse tasks. Notably, it achieves robust zero-shot performance in generating universal cell representations and exhibits excellent transferability in cell type annotation and cross-omics prediction. By uncovering enhancer-gene links undetected by existing computational methods, ChromFound offers a promising framework for understanding disease risk variants in the noncoding genome.
Single-cell RNA sequencing (scRNA-seq) reveals cell heterogeneity, with cell clustering playing a key role in identifying cell types and marker genes. Recent advances, especially graph neural networks (GNNs)-based methods, have significantly improved clustering performance. However, the analysis of scRNA-seq data remains challenging due to noise, sparsity, and high dimensionality. Compounding these challenges, GNNs often suffer from over-smoothing, limiting their ability to capture complex biological information. In response, we propose scSiameseClu, a novel Siamese Clustering framework for interpreting single-cell RNA-seq data, comprising of 3 key steps: (1) Dual Augmentation Module, which applies biologically informed perturbations to the gene expression matrix and cell graph relationships to enhance representation robustness; (2) Siamese Fusion Module, which combines cross-correlation refinement and adaptive information fusion to capture complex cellular relationships while mitigating over-smoothing; and (3) Optimal Transport Clustering, which utilizes Sinkhorn distance to efficiently align cluster assignments with predefined proportions while maintaining balance. Comprehensive evaluations on seven real-world datasets demonstrate that scSiameseClu outperforms state-of-the-art methods in single-cell clustering, cell type annotation, and cell type classification, providing a powerful tool for scRNA-seq data interpretation.
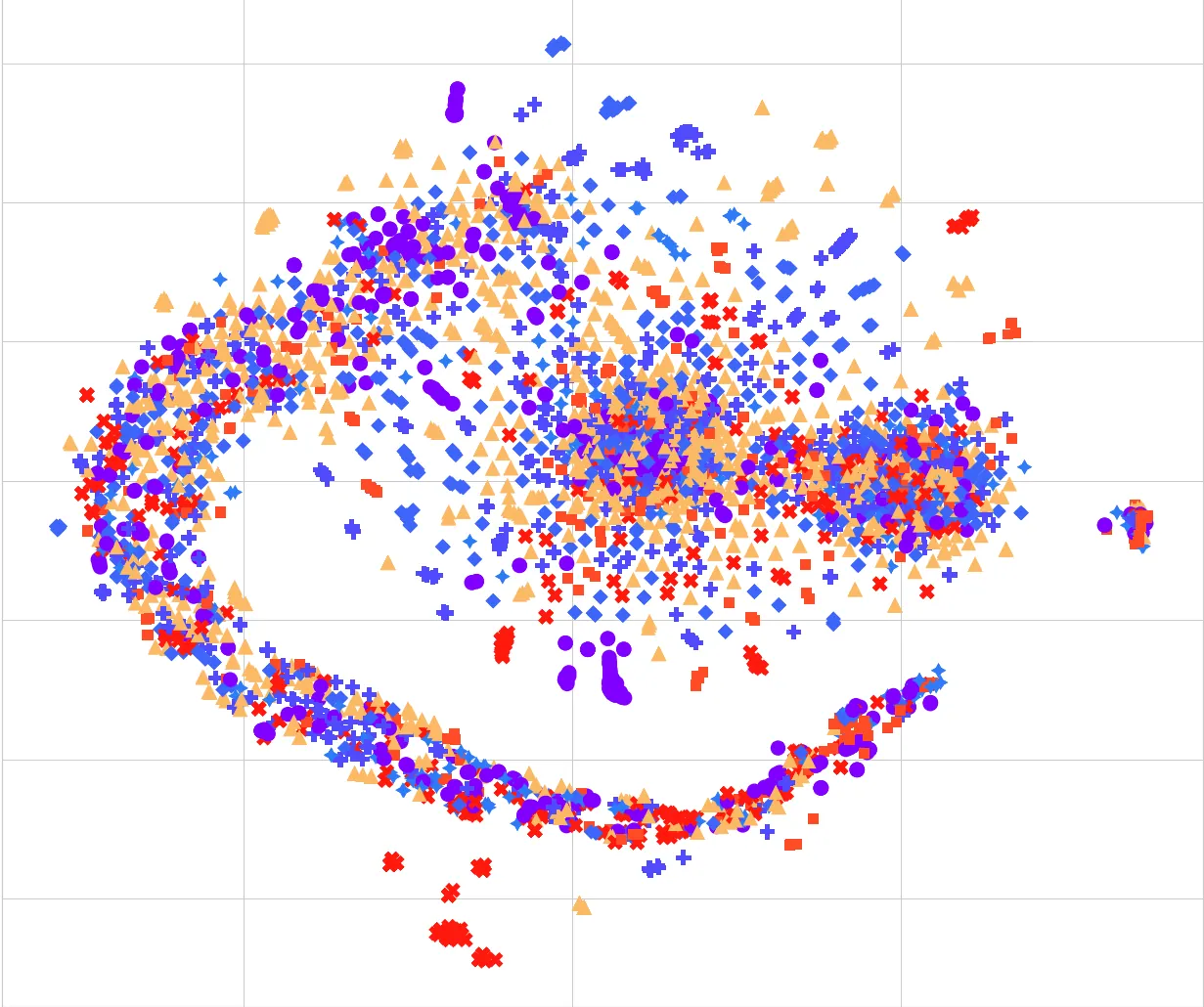
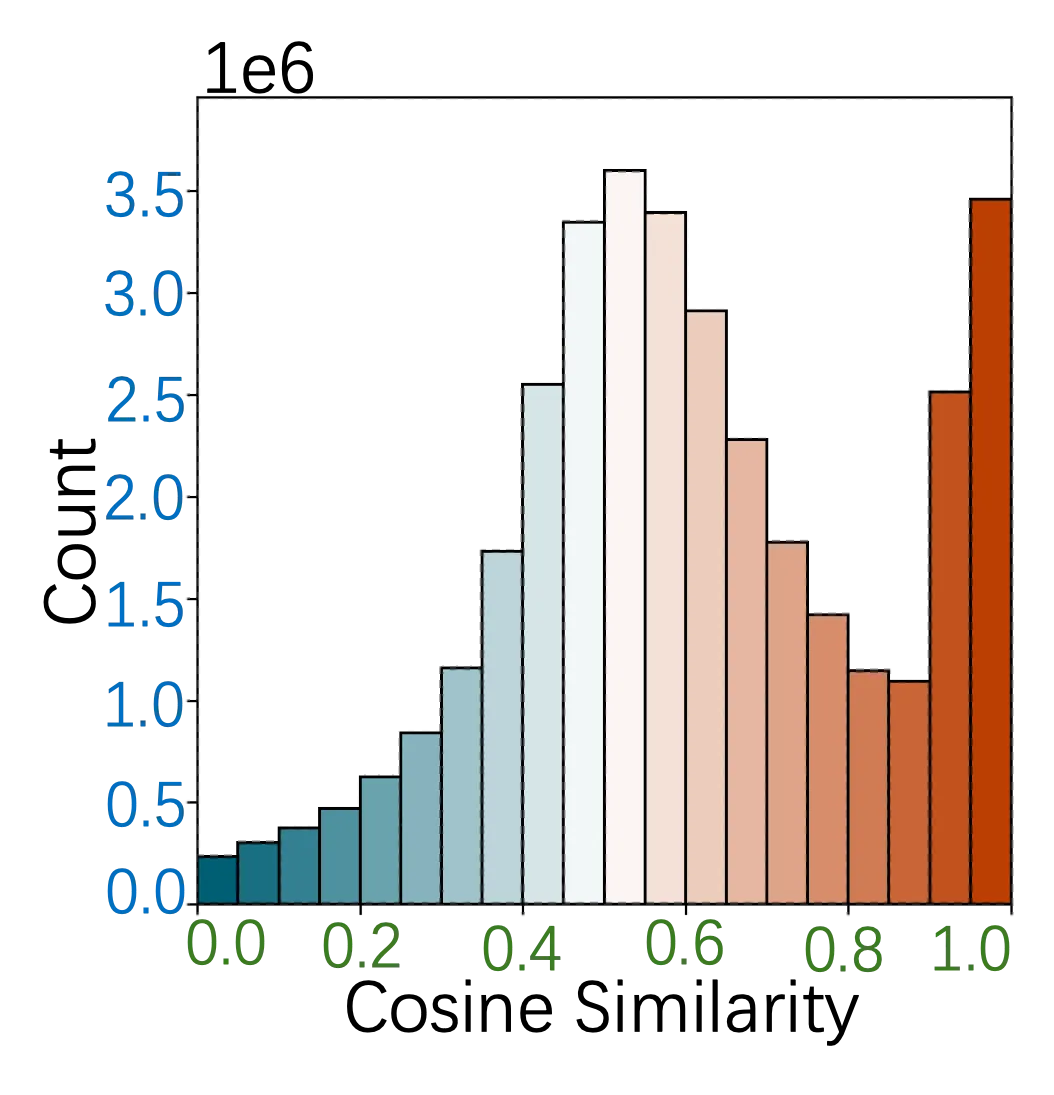
Molecular sequence analysis is crucial for comprehending several biological processes, including protein-protein interactions, functional annotation, and disease classification. The large number of sequences and the inherently complicated nature of protein structures make it challenging to analyze such data. Finding patterns and enhancing subsequent research requires the use of dimensionality reduction and feature selection approaches. Recently, a method called Correlated Clustering and Projection (CCP) has been proposed as an effective method for biological sequencing data. The CCP technique is still costly to compute even though it is effective for sequence visualization. Furthermore, its utility for classifying molecular sequences is still uncertain. To solve these two problems, we present a Nearest Neighbor Correlated Clustering and Projection (CCP-NN)-based technique for efficiently preprocessing molecular sequence data. To group related molecular sequences and produce representative supersequences, CCP makes use of sequence-to-sequence correlations. As opposed to conventional methods, CCP doesn't rely on matrix diagonalization, therefore it can be applied to a range of machine-learning problems. We estimate the density map and compute the correlation using a nearest-neighbor search technique. We performed molecular sequence classification using CCP and CCP-NN representations to assess the efficacy of our proposed approach. Our findings show that CCP-NN considerably improves classification task accuracy as well as significantly outperforms CCP in terms of computational runtime.
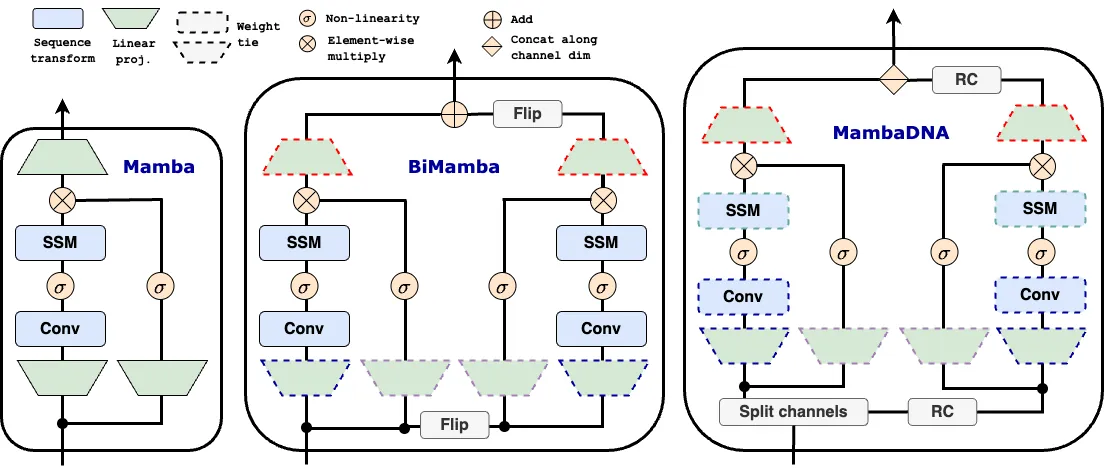
Large-scale sequence modeling has sparked rapid advances that now extend into biology and genomics. However, modeling genomic sequences introduces challenges such as the need to model long-range token interactions, the effects of upstream and downstream regions of the genome, and the reverse complementarity (RC) of DNA. Here, we propose an architecture motivated by these challenges that builds off the long-range Mamba block, and extends it to a BiMamba component that supports bi-directionality, and to a MambaDNA block that additionally supports RC equivariance. We use MambaDNA as the basis of Caduceus, the first family of RC equivariant bi-directional long-range DNA language models, and we introduce pre-training and fine-tuning strategies that yield Caduceus DNA foundation models. Caduceus outperforms previous long-range models on downstream benchmarks; on a challenging long-range variant effect prediction task, Caduceus exceeds the performance of 10x larger models that do not leverage bi-directionality or equivariance.
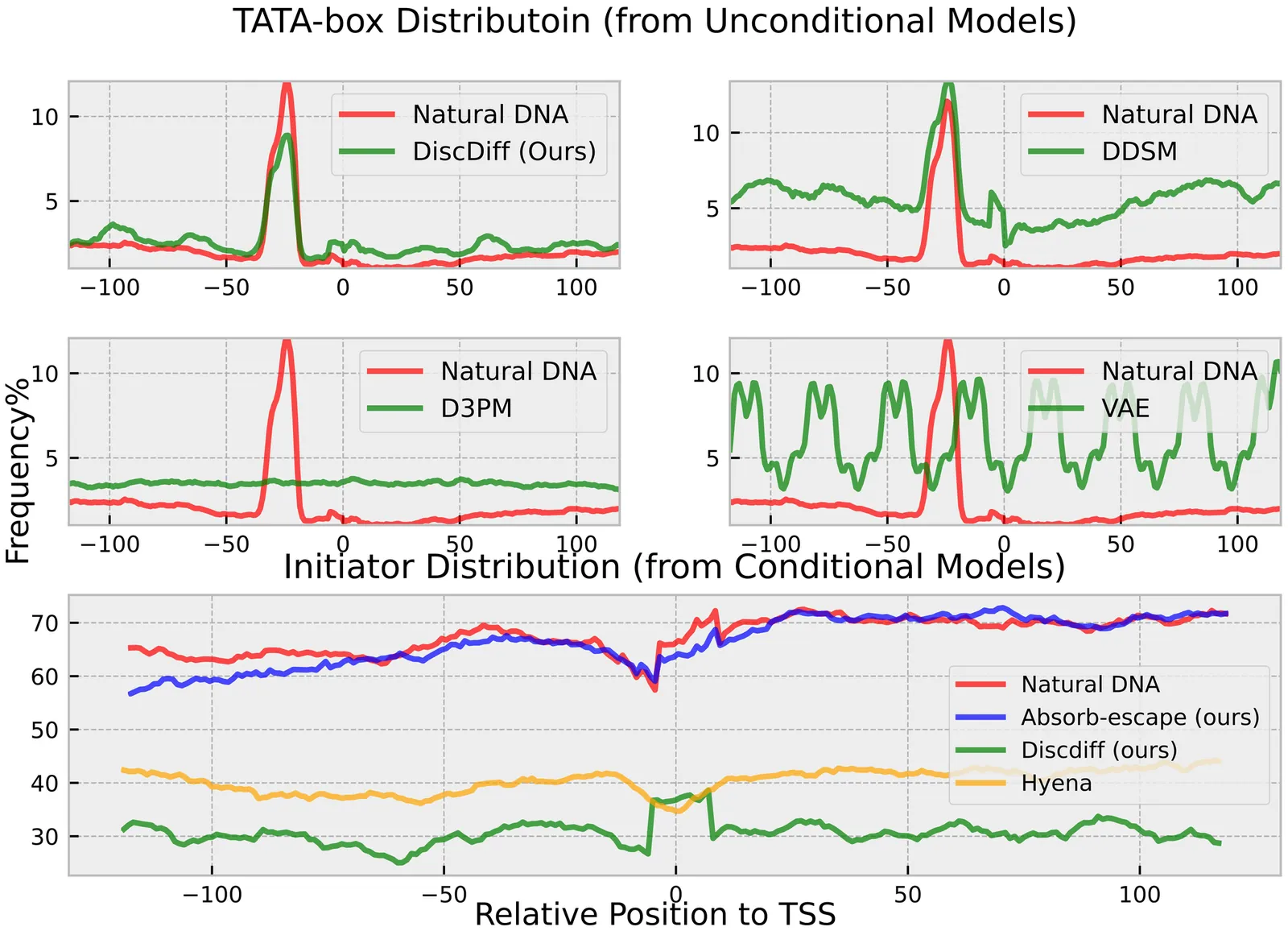
This paper introduces a novel framework for DNA sequence generation, comprising two key components: DiscDiff, a Latent Diffusion Model (LDM) tailored for generating discrete DNA sequences, and Absorb-Escape, a post-training algorithm designed to refine these sequences. Absorb-Escape enhances the realism of the generated sequences by correcting `round errors' inherent in the conversion process between latent and input spaces. Our approach not only sets new standards in DNA sequence generation but also demonstrates superior performance over existing diffusion models, in generating both short and long DNA sequences. Additionally, we introduce EPD-GenDNA, the first comprehensive, multi-species dataset for DNA generation, encompassing 160,000 unique sequences from 15 species. We hope this study will advance the generative modelling of DNA, with potential implications for gene therapy and protein production.

Language models, especially transformer-based ones, have achieved colossal success in NLP. To be precise, studies like BERT for NLU and works like GPT-3 for NLG are very important. If we consider DNA sequences as a text written with an alphabet of four letters representing the nucleotides, they are similar in structure to natural languages. This similarity has led to the development of discriminative language models such as DNABert in the field of DNA-related bioinformatics. To our knowledge, however, the generative side of the coin is still largely unexplored. Therefore, we have focused on the development of an autoregressive generative language model such as GPT-3 for DNA sequences. Since working with whole DNA sequences is challenging without extensive computational resources, we decided to conduct our study on a smaller scale and focus on nucleotide sequences of human genes rather than the whole DNA. This decision has not changed the structure of the problem, as both DNA and genes can be considered as 1D sequences consisting of four different nucleotides without losing much information and without oversimplification. Firstly, we systematically studied an almost entirely unexplored problem and observed that RNNs perform best, while simple techniques such as N-grams are also promising. Another beneficial point was learning how to work with generative models on languages we do not understand, unlike natural languages. The importance of using real-world tasks beyond classical metrics such as perplexity was noted. In addition, we examined whether the data-hungry nature of these models can be altered by selecting a language with minimal vocabulary size, four due to four different types of nucleotides. The reason for reviewing this was that choosing such a language might make the problem easier. However, in this study, we found that this did not change the amount of data required very much.

Decoding the linguistic intricacies of the genome is a crucial problem in biology, and pre-trained foundational models such as DNABERT and Nucleotide Transformer have made significant strides in this area. Existing works have largely hinged on k-mer, fixed-length permutations of A, T, C, and G, as the token of the genome language due to its simplicity. However, we argue that the computation and sample inefficiencies introduced by k-mer tokenization are primary obstacles in developing large genome foundational models. We provide conceptual and empirical insights into genome tokenization, building on which we propose to replace k-mer tokenization with Byte Pair Encoding (BPE), a statistics-based data compression algorithm that constructs tokens by iteratively merging the most frequent co-occurring genome segment in the corpus. We demonstrate that BPE not only overcomes the limitations of k-mer tokenization but also benefits from the computational efficiency of non-overlapping tokenization. Based on these insights, we introduce DNABERT-2, a refined genome foundation model that adapts an efficient tokenizer and employs multiple strategies to overcome input length constraints, reduce time and memory expenditure, and enhance model capability. Furthermore, we identify the absence of a comprehensive and standardized benchmark for genome understanding as another significant impediment to fair comparative analysis. In response, we propose the Genome Understanding Evaluation (GUE), a comprehensive multi-species genome classification dataset that amalgamates $36$ distinct datasets across $9$ tasks, with input lengths ranging from $70$ to $10000$. Through comprehensive experiments on the GUE benchmark, we demonstrate that DNABERT-2 achieves comparable performance to the state-of-the-art model with $21 \times$ fewer parameters and approximately $92 \times$ less GPU time in pre-training.
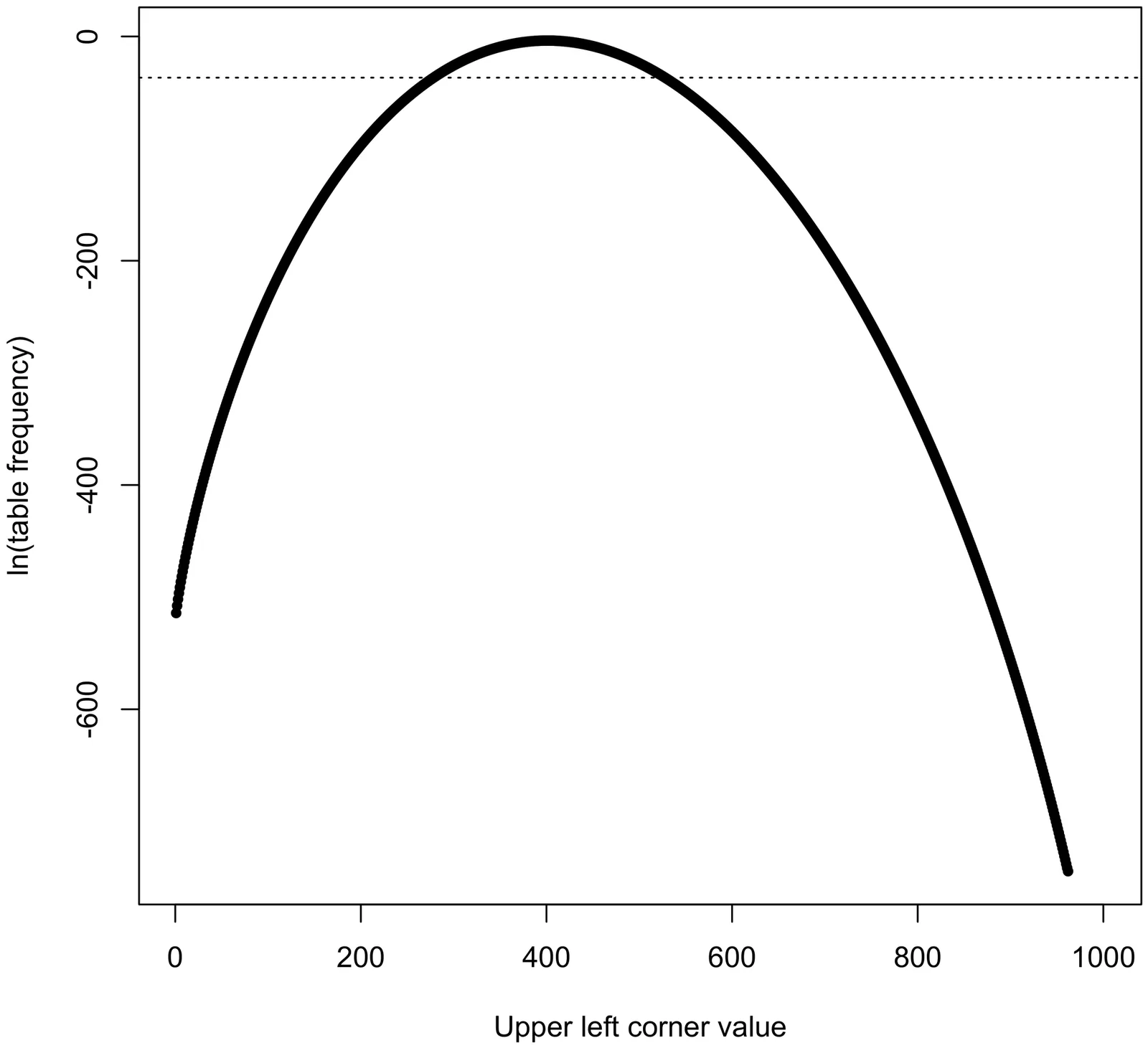
PLINK 1 is a widely used open-source C/C++ toolset for genome-wide association studies (GWAS) and research in population genetics. However, the steady accumulation of data from imputation and whole-genome sequencing studies has exposed a strong need for even faster and more scalable implementations of key functions. In addition, GWAS and population-genetic data now frequently contain probabilistic calls, phase information, and/or multiallelic variants, none of which can be represented by PLINK 1's primary data format. To address these issues, we are developing a second-generation codebase for PLINK. The first major release from this codebase, PLINK 1.9, introduces extensive use of bit-level parallelism, O(sqrt(n))-time/constant-space Hardy-Weinberg equilibrium and Fisher's exact tests, and many other algorithmic improvements. In combination, these changes accelerate most operations by 1-4 orders of magnitude, and allow the program to handle datasets too large to fit in RAM. This will be followed by PLINK 2.0, which will introduce (a) a new data format capable of efficiently representing probabilities, phase, and multiallelic variants, and (b) extensions of many functions to account for the new types of information. The second-generation versions of PLINK will offer dramatic improvements in performance and compatibility. For the first time, users without access to high-end computing resources can perform several essential analyses of the feature-rich and very large genetic datasets coming into use.