Trending in Computational Biology
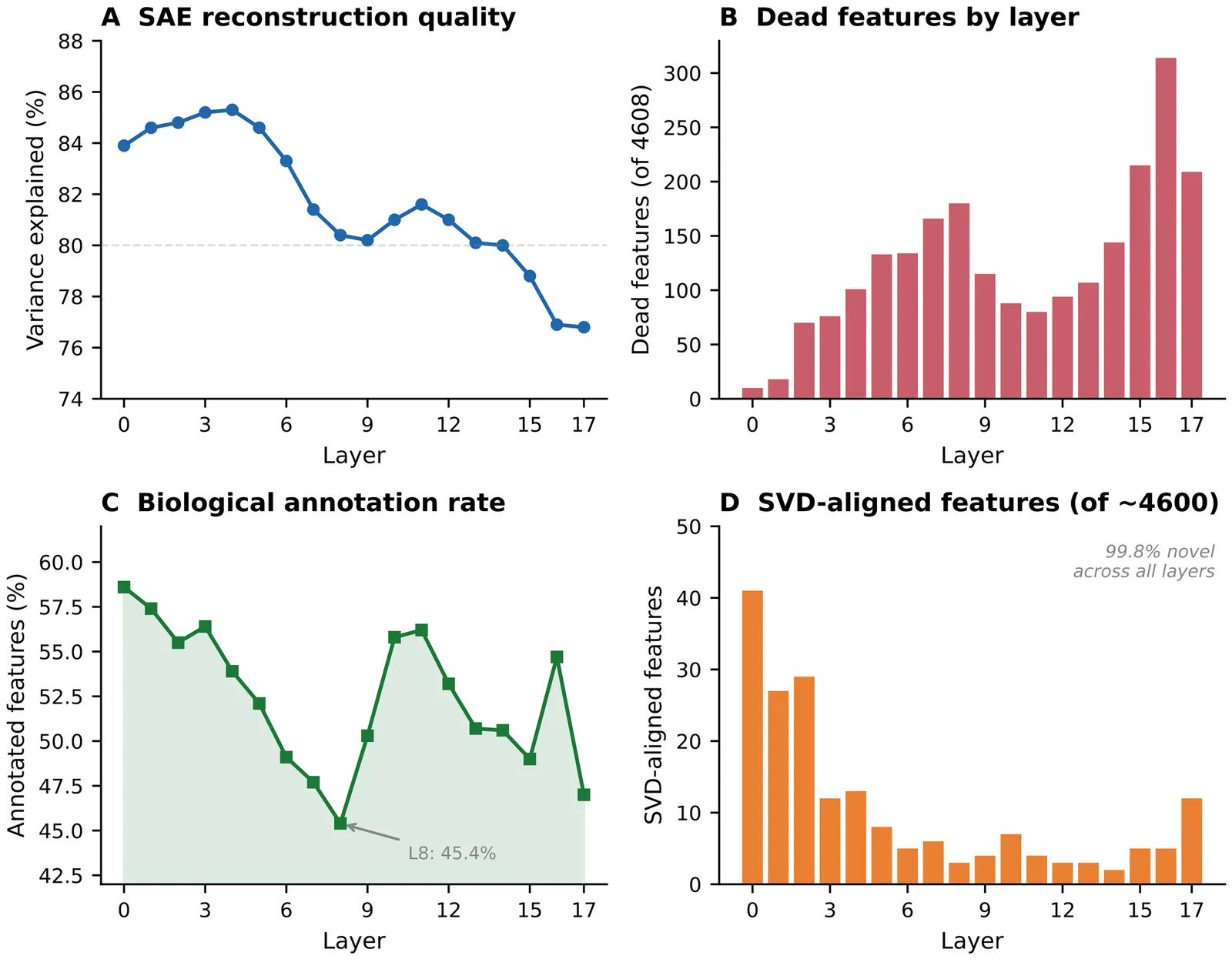
Sparse autoencoders reveal organized biological knowledge but minimal regulatory logic in single-cell foundation models: a comparative atlas of Geneformer and scGPT
Background: Single-cell foundation models such as Geneformer and scGPT encode rich biological information, but whether this includes causal regulatory logic rather than statistical co-expression remains unclear. Sparse autoencoders (SAEs) can resolve superposition in neural networks by decomposing dense activations into interpretable features, yet they have not been systematically applied to biological foundation models. Results: We trained TopK SAEs on residual stream activations from all layers of Geneformer V2-316M (18 layers, d=1152) and scGPT whole-human (12 layers, d=512), producing atlases of 82525 and 24527 features, respectively. Both atlases confirm massive superposition, with 99.8 percent of features invisible to SVD. Systematic characterization reveals rich biological organization: 29 to 59 percent of features annotate to Gene Ontology, KEGG, Reactome, STRING, or TRRUST, with U-shaped layer profiles reflecting hierarchical abstraction. Features organize into co-activation modules (141 in Geneformer, 76 in scGPT), exhibit causal specificity (median 2.36x), and form cross-layer information highways (63 to 99.8 percent). When tested against genome-scale CRISPRi perturbation data, only 3 of 48 transcription factors (6.2 percent) show regulatory-target-specific feature responses. A multi-tissue control yields marginal improvement (10.4 percent, 5 of 48 TFs), establishing model representations as the bottleneck. Conclusions: These models have internalized organized biological knowledge, including pathway membership, protein interactions, functional modules, and hierarchical abstraction, yet they encode minimal causal regulatory logic. We release both feature atlases as interactive web platforms enabling exploration of more than 107000 features across 30 layers of two leading single-cell foundation models.
2603.029521
Mar 2026Genomics
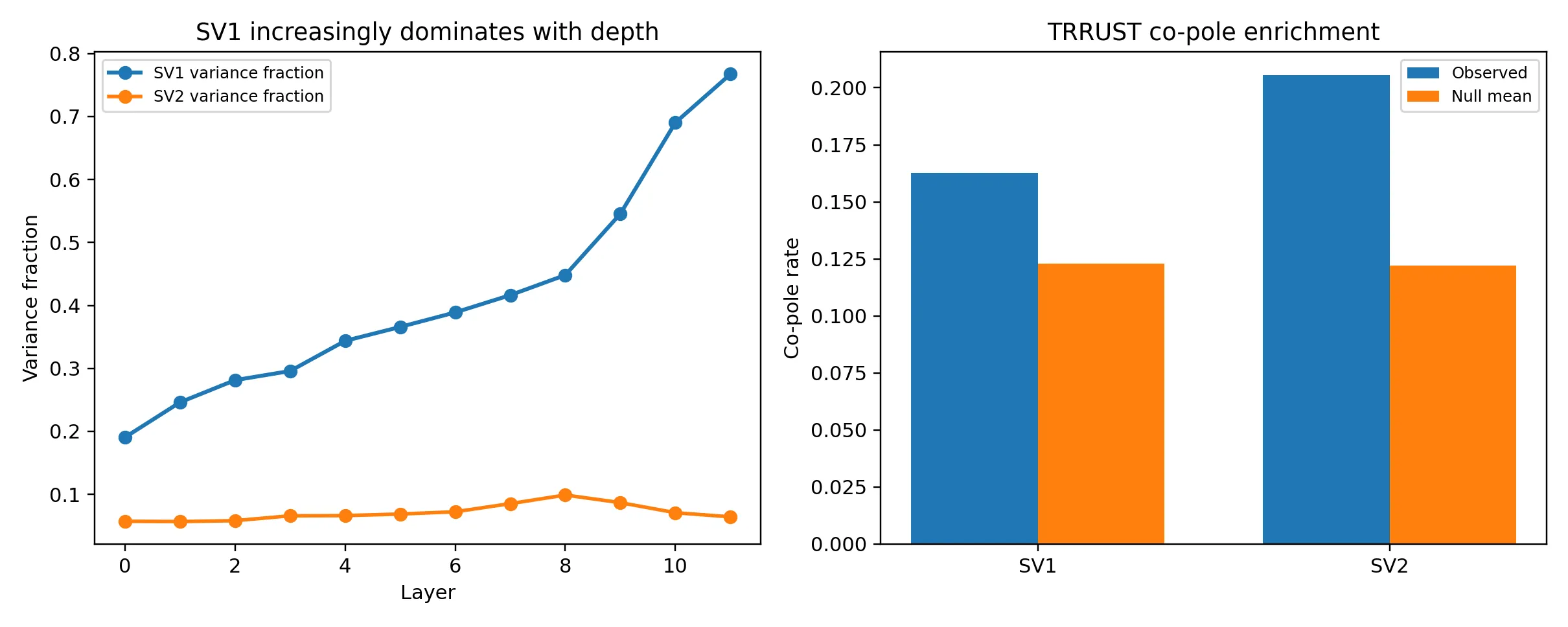
Multi-Dimensional Spectral Geometry of Biological Knowledge in Single-Cell Transformer Representations
Single-cell foundation models such as scGPT learn high-dimensional gene representations, but what biological knowledge these representations encode remains unclear. We systematically decode the geometric structure of scGPT internal representations through 63 iterations of automated hypothesis screening (183 hypotheses tested), revealing that the model organizes genes into a structured biological coordinate system rather than an opaque feature space. The dominant spectral axis separates genes by subcellular localization, with secreted proteins at one pole and cytosolic proteins at the other. Intermediate transformer layers transiently encode mitochondrial and ER compartments in a sequence that mirrors the cellular secretory pathway. Orthogonal axes encode protein-protein interaction networks with graded fidelity to experimentally measured interaction strength (Spearman rho = 1.000 across n = 5 STRING confidence quintiles, p = 0.017). In a compact six-dimensional spectral subspace, the model distinguishes transcription factors from their target genes (AUROC = 0.744, all 12 layers significant). Early layers preserve which specific genes regulate which targets, while deeper layers compress this into a coarser regulator versus regulated distinction. Repression edges are geometrically more prominent than activation edges, and B-cell master regulators BATF and BACH2 show convergence toward the B-cell identity anchor PAX5 across transformer depth. Cell-type marker genes cluster with high fidelity (AUROC = 0.851). Residual-stream geometry encodes biological structure complementary to attention patterns. These results indicate that biological transformers learn an interpretable internal model of cellular organization, with implications for regulatory network inference, drug target prioritization, and model auditing.
2602.222471
Feb 2026Genomics
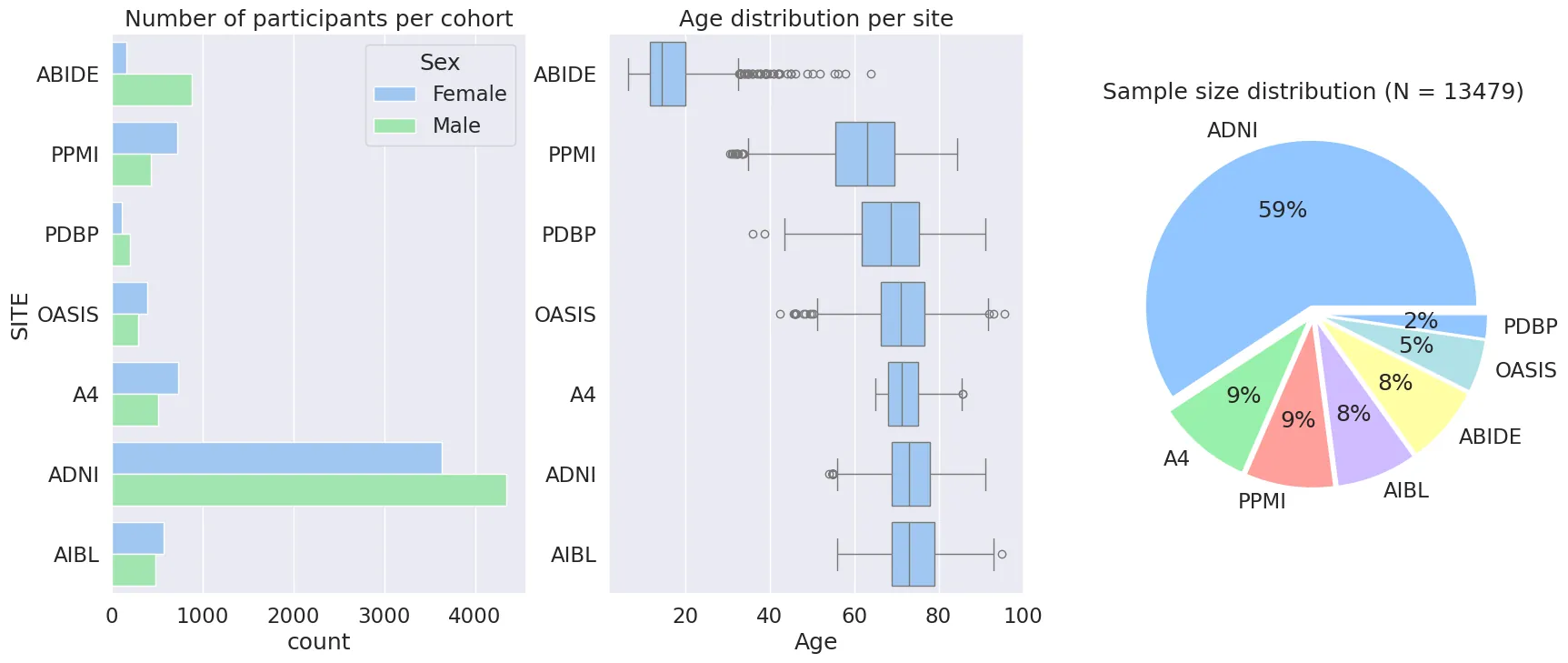
Fed-ComBat: A Generalized Federated Framework for Batch Effect Harmonization in Collaborative Studies
The use of multi-centric analyses is crucial for obtaining sufficient sample sizes and representative clinical populations in experimental studies. In this setting, data harmonization techniques are typically employed to address systematic biases and ensure the interoperability of the data. State-of-the-art harmonisation approaches are based on the statistical theory of random effect modeling, allowing to account for either linear of non-linear biases and batch effects. However, optimizing these statistical methods generally requires data centralization at some point during the analysis pipeline, therefore introducing the risk of exposing individual patient information while posing significant data governance issues. To overcome this challenge, in this paper we present Fed-ComBat, a federated framework for batch effect harmonization on decentralized data. Fed-ComBat enables the preservation of nonlinear covariate effects without requiring centralization of data and without prior parametric hypothesis on the variables to account for. We demonstrate the effectiveness of Fed-ComBat against a comprehensive panel of existing approaches based on the state-of-the-art ComBat, along with distributed and nonlinear variants. Our experiments are based on extensive simulated data, and on the analysis of multiple cohorts based on 7 neuroimaging studies comprising healthy controls (CI) and subjects with various disorders such as Parkinson's disease (PD), Alzheimer's disease (AD), and autism spectrum disorder (ASD). Our results show that in a federated settings, Fed-ComBat harmonization exhibits comparable results to centralized methods for both linear and nonlinear cases. On real data, harmonized trajectories of the thickness ofthe right hippocampus across lifespan measured on a set of 7 public studies show comparable results between centralized and federated models and are consistent with the literature when using a nonlinear model. The code is publicly available at: https://gitlab.inria.fr/greguig/fedcombat
2601.143142
Jan 2026Quantitative Methods
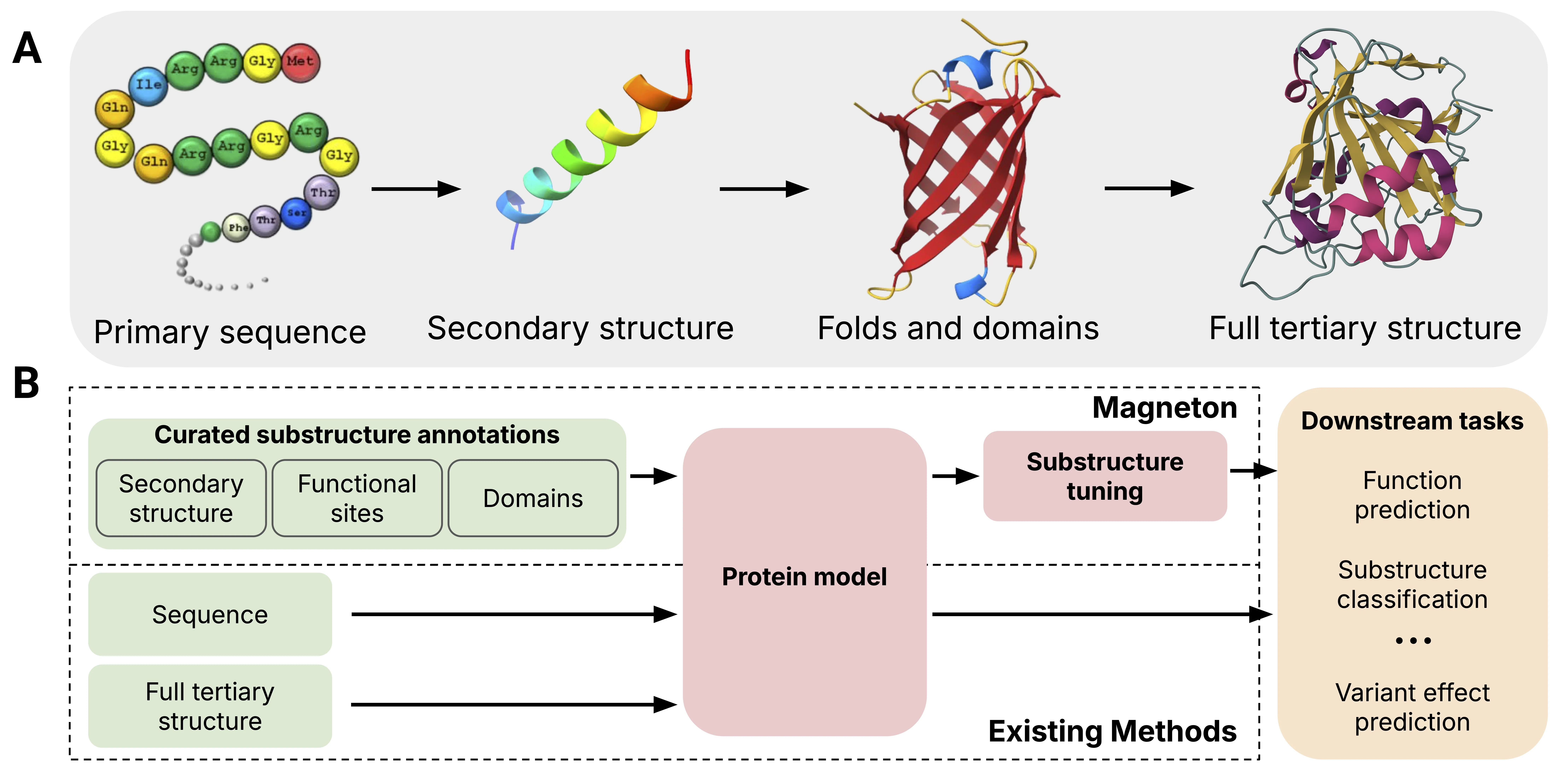
Greater than the Sum of Its Parts: Building Substructure into Protein Encoding Models
Protein representation learning has advanced rapidly with the scale-up of sequence and structure supervision, but most models still encode proteins either as per-residue token sequences or as single global embeddings. This overlooks a defining property of protein organization: proteins are built from recurrent, evolutionarily conserved substructures that concentrate biochemical activity and mediate core molecular functions. Although substructures such as domains and functional sites are systematically cataloged, they are rarely used as training signals or representation units in protein models. We introduce Magneton, an environment for developing substructure-aware protein models. Magneton provides (1) a dataset of 530,601 proteins annotated with over 1.7 million substructures spanning 13,075 types, (2) a training framework for incorporating substructures into existing protein models, and (3) a benchmark suite of 13 tasks probing representations at the residue, substructural, and protein levels. Using Magneton, we develop substructure-tuning, a supervised fine-tuning method that distills substructural knowledge into pretrained protein models. Across state-of-the-art sequence- and structure-based models, substructure-tuning improves function prediction, yields more consistent representations of substructure types never observed during tuning, and shows that substructural supervision provides information that is complementary to global structure inputs. The Magneton environment, datasets, and substructure-tuned models are all openly available (https://github.com/rcalef/magneton/).
2512.181141
Dec 2025Quantitative Methods
The TAG array of a multiple sequence alignment
Modern genomic analyses increasingly rely on pangenomes, that is, representations of the genome of entire populations. The simplest representation of a pangenome is a set of individual genome sequences. Compared to e.g. sequence graphs, this has the advantage that efficient exact search via indexes based on the Burrows-Wheeler Transform (BWT) is possible, that no chimeric sequences are created, and that the results are not influenced by heuristics. However, such an index may report a match in thousands of positions even if these all correspond to the same locus, making downstream analysis unnecessarily expensive. For sufficiently similar sequences (e.g. human chromosomes), a multiple sequence alignment (MSA) can be computed. Since an MSA tends to group similar strings in the same columns, it is likely that a string occurring thousands of times in the pangenome can be described by very few columns in the MSA. We describe a method to tag entries in the BWT with the corresponding column in the MSA and develop an index that can map matches in the BWT to columns in the MSA in time proportional to the output. As a by-product, we can efficiently project a match to a designated reference genome, a capability that current pangenome aligners based on the BWT lack.
2511.190681
Nov 2025Genomics
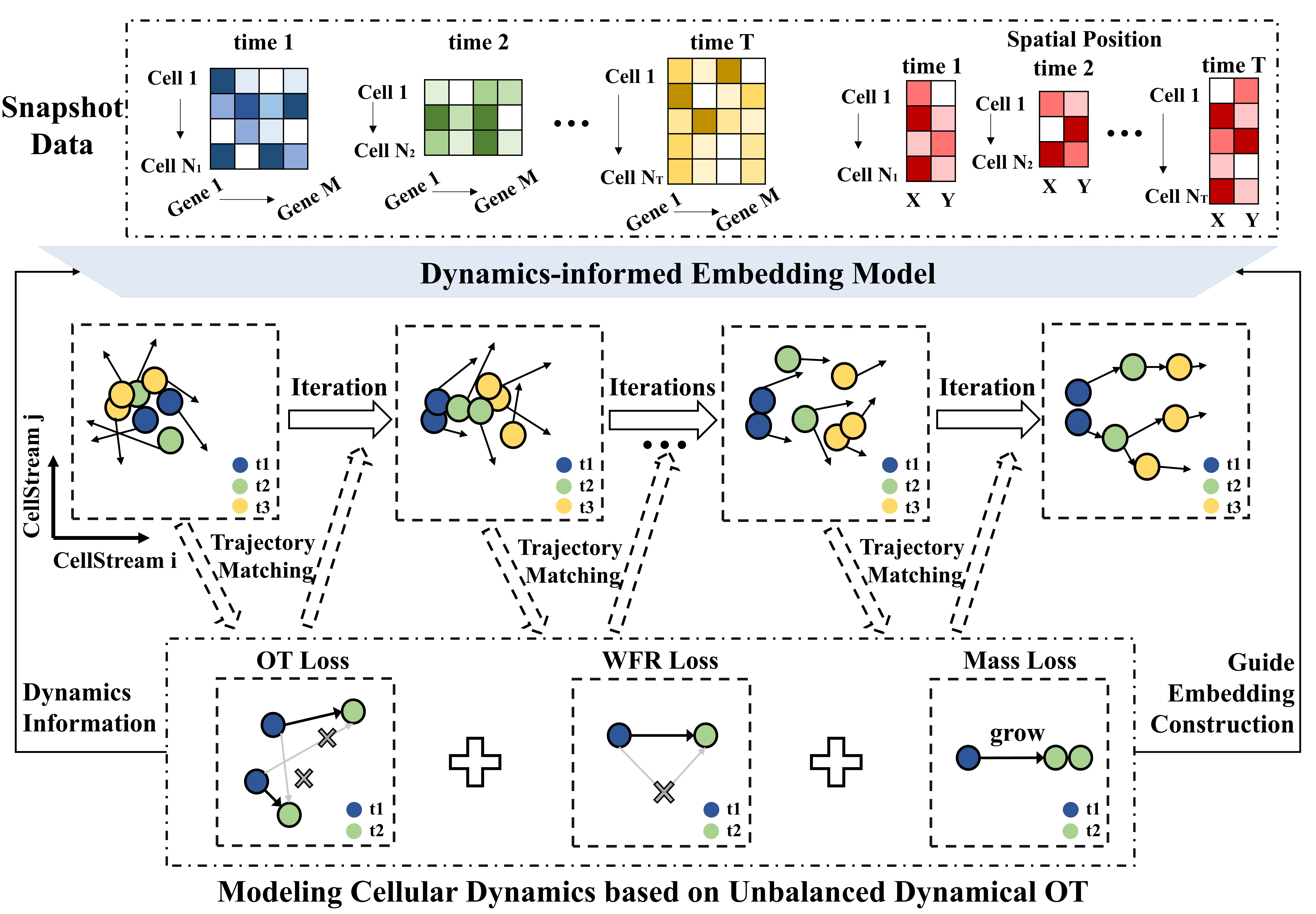
CellStream: Dynamical Optimal Transport Informed Embeddings for Reconstructing Cellular Trajectories from Snapshots Data
Single-cell RNA sequencing (scRNA-seq), especially temporally resolved datasets, enables genome-wide profiling of gene expression dynamics at single-cell resolution across discrete time points. However, current technologies provide only sparse, static snapshots of cell states and are inherently influenced by technical noise, complicating the inference and representation of continuous transcriptional dynamics. Although embedding methods can reduce dimensionality and mitigate technical noise, the majority of existing approaches typically treat trajectory inference separately from embedding construction, often neglecting temporal structure. To address this challenge, here we introduce CellStream, a novel deep learning framework that jointly learns embedding and cellular dynamics from single-cell snapshot data by integrating an autoencoder with unbalanced dynamical optimal transport. Compared to existing methods, CellStream generates dynamics-informed embeddings that robustly capture temporal developmental processes while maintaining high consistency with the underlying data manifold. We demonstrate CellStream's effectiveness on both simulated datasets and real scRNA-seq data, including spatial transcriptomics. Our experiments indicate significant quantitative improvements over state-of-the-art methods in representing cellular trajectories with enhanced temporal coherence and reduced noise sensitivity. Overall, CellStream provides a new tool for learning and representing continuous streams from the noisy, static snapshots of single-cell gene expression.
2511.137861
Nov 2025Genomics
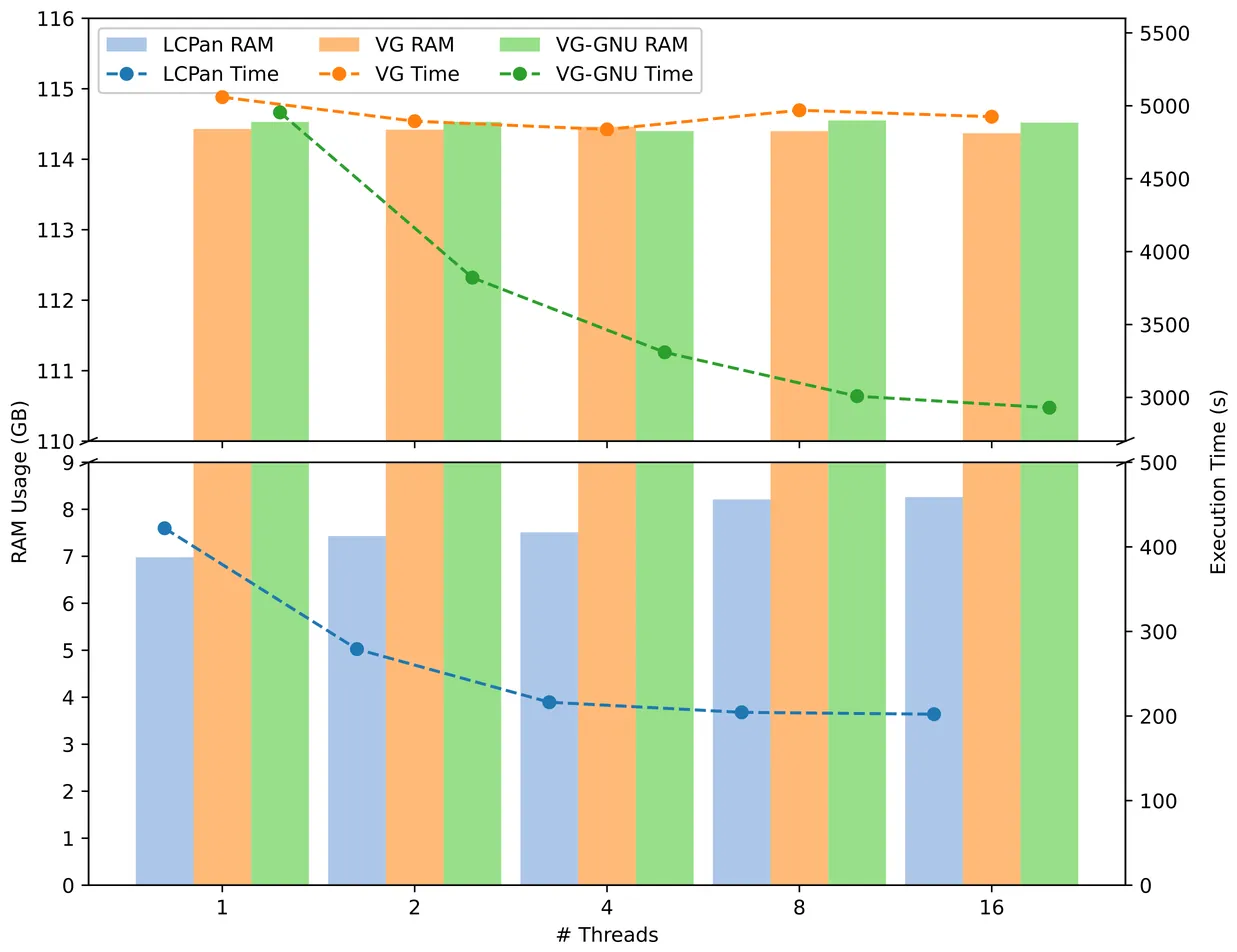
LCPan: efficient variation graph construction using Locally Consistent Parsing
Efficient and consistent string processing is critical in the exponentially growing genomic data era. Locally Consistent Parsing (LCP) addresses this need by partitioning an input genome string into short, exactly matching substrings (e.g., "cores"), ensuring consistency across partitions. Labeling the cores of an input string consistently not only provides a compact representation of the input but also enables the reapplication of LCP to refine the cores over multiple iterations, providing a progressively longer and more informative set of substrings for downstream analyses. We present the first iterative implementation of LCP with Lcptools and demonstrate its effectiveness in identifying cores with minimal collisions. Experimental results show that the number of cores at the i^th iteration is O(n/c^i) for c ~ 2.34, while the average length and the average distance between consecutive cores are O(c^i). Compared to the popular sketching techniques, LCP produces significantly fewer cores, enabling a more compact representation and faster analyses. To demonstrate the advantages of LCP in genomic string processing in terms of computation and memory efficiency, we also introduce LCPan, an efficient variation graph constructor. We show that LCPan generates variation graphs >10x faster than vg, while using >13x less memory.
2511.122051
Nov 2025Genomics

Protein Structure Tokenization via Geometric Byte Pair Encoding
Protein structure is central to biological function, and enabling multimodal protein models requires joint reasoning over sequence, structure, and function. A key barrier is the lack of principled protein structure tokenizers (PSTs): existing approaches fix token size or rely on continuous vector codebooks, limiting interpretability, multi-scale control, and transfer across architectures. We introduce GeoBPE, a geometry-grounded PST that transforms continuous, noisy, multi-scale backbone conformations into discrete ``sentences'' of geometry while enforcing global constraints. Analogous to byte-pair encoding, GeoBPE generates a hierarchical vocabulary of geometric primitives by iteratively (i) clustering Geo-Pair occurrences with k-medoids to yield a resolution-controllable vocabulary; (ii) quantizing each Geo-Pair to its closest medoid prototype; and (iii) reducing drift through differentiable inverse kinematics that optimizes boundary glue angles under an $\mathrm{SE}(3)$ end-frame loss. GeoBPE offers compression ($>$10x reduction in bits-per-residue at similar distortion rate), data efficiency ($>$10x less training data), and generalization (maintains test/train distortion ratio of $1.0-1.1$). It is architecture-agnostic: (a) its hierarchical vocabulary provides a strong inductive bias for coarsening residue-level embeddings from large PLMs into motif- and protein-level representations, consistently outperforming leading PSTs across $12$ tasks and $24$ test splits; (b) paired with a transformer, GeoBPE supports unconditional backbone generation via language modeling; and (c) tokens align with CATH functional families and support expert-interpretable case studies, offering functional meaning absent in prior PSTs. Code is available at https://github.com/shiningsunnyday/PT-BPE/.
2511.117581
Nov 2025Quantitative Methods
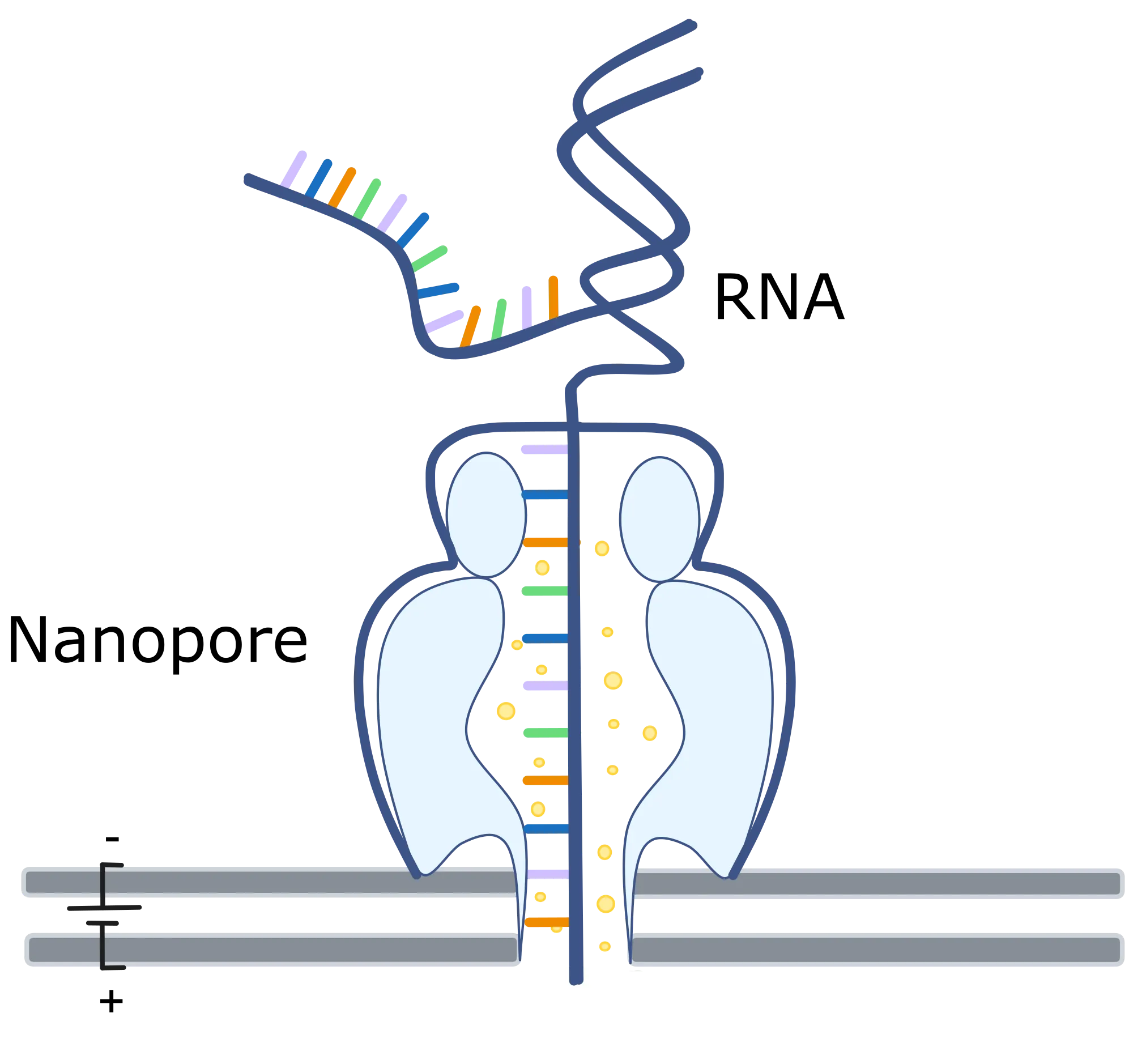
Path Signatures Enable Model-Free Mapping of RNA Modifications
Detecting chemical modifications on RNA molecules remains a key challenge in epitranscriptomics. Traditional reverse transcription-based sequencing methods introduce enzyme- and sequence-dependent biases and fragment RNA molecules, confounding the accurate mapping of modifications across the transcriptome. Nanopore direct RNA sequencing offers a powerful alternative by preserving native RNA molecules, enabling the detection of modifications at single-molecule resolution. However, current computational tools can identify only a limited subset of modification types within well-characterized sequence contexts for which ample training data exists. Here, we introduce a model-free computational method that reframes modification detection as an anomaly detection problem, requiring only canonical (unmodified) RNA reads without any other annotated data. For each nanopore read, our approach extracts robust, modification-sensitive features from the raw ionic current signal at a site using the signature transform, then computes an anomaly score by comparing the resulting feature vector to its nearest neighbors in an unmodified reference dataset. We convert anomaly scores into statistical p-values to enable anomaly detection at both individual read and site levels. Validation on densely-modified \textit{E. coli} rRNA demonstrates that our approach detects known sites harboring diverse modification types, without prior training on these modifications. We further applyied this framework to dengue virus (DENV) transcripts and mammalian mRNAs. For DENV sfRNA, it led to revealing a novel 2'-O-methylated site, which we validate orthogonally by qRT-PCR assays. These results demonstrate that our model-free approach operates robustly across different types of RNAs and datasets generated with different nanopore sequencing chemistries.
2511.088551
Nov 2025Genomics

Fast, memory-efficient genomic interval tokenizers for modern machine learning
Introduction: Epigenomic datasets from high-throughput sequencing experiments are commonly summarized as genomic intervals. As the volume of this data grows, so does interest in analyzing it through deep learning. However, the heterogeneity of genomic interval data, where each dataset defines its own regions, creates barriers for machine learning methods that require consistent, discrete vocabularies. Methods: We introduce gtars-tokenizers, a high-performance library that maps genomic intervals to a predefined universe or vocabulary of regions, analogous to text tokenization in natural language processing. Built in Rust with bindings for Python, R, CLI, and WebAssembly, gtars-tokenizers implements two overlap methods (BITS and AIList) and integrates seamlessly with modern ML frameworks through Hugging Face-compatible APIs. Results: The gtars-tokenizers package achieves top efficiency for large-scale datasets, while enabling genomic intervals to be processed using standard ML workflows in PyTorch and TensorFlow without ad hoc preprocessing. This token-based approach bridges genomics and machine learning, supporting scalable and standardized analysis of interval data across diverse computational environments. Availability: PyPI and GitHub: https://github.com/databio/gtars.
2511.015551
Nov 2025Genomics
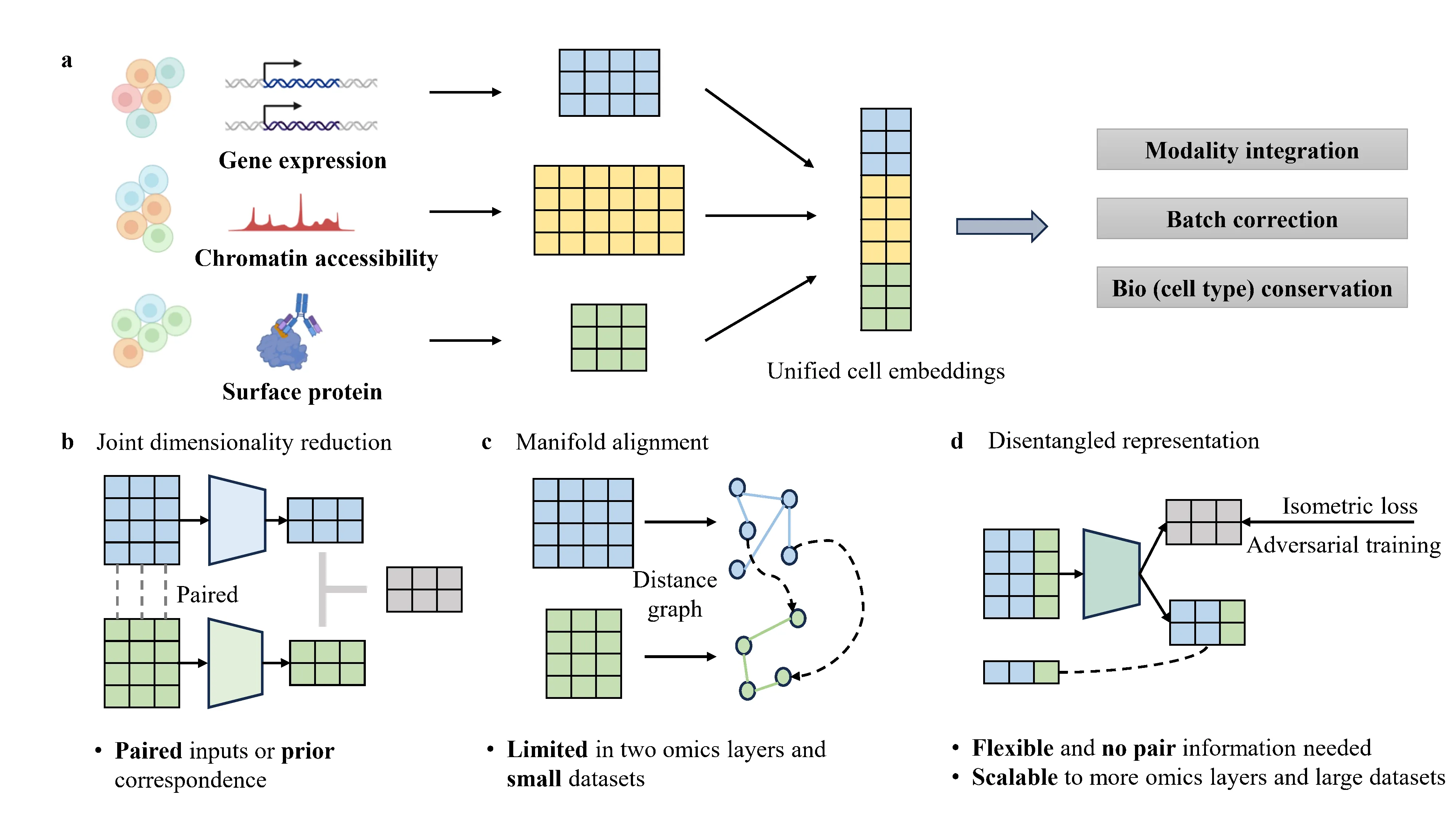
scMRDR: A scalable and flexible framework for unpaired single-cell multi-omics data integration
Advances in single-cell sequencing have enabled high-resolution profiling of diverse molecular modalities, while integrating unpaired multi-omics single-cell data remains challenging. Existing approaches either rely on pair information or prior correspondences, or require computing a global pairwise coupling matrix, limiting their scalability and flexibility. In this paper, we introduce a scalable and flexible generative framework called single-cell Multi-omics Regularized Disentangled Representations (scMRDR) for unpaired multi-omics integration. Specifically, we disentangle each cell's latent representations into modality-shared and modality-specific components using a well-designed $β$-VAE architecture, which are augmented with isometric regularization to preserve intra-omics biological heterogeneity, adversarial objective to encourage cross-modal alignment, and masked reconstruction loss strategy to address the issue of missing features across modalities. Our method achieves excellent performance on benchmark datasets in terms of batch correction, modality alignment, and biological signal preservation. Crucially, it scales effectively to large-level datasets and supports integration of more than two omics, offering a powerful and flexible solution for large-scale multi-omics data integration and downstream biological discovery.
2510.249872
Oct 2025Quantitative Methods
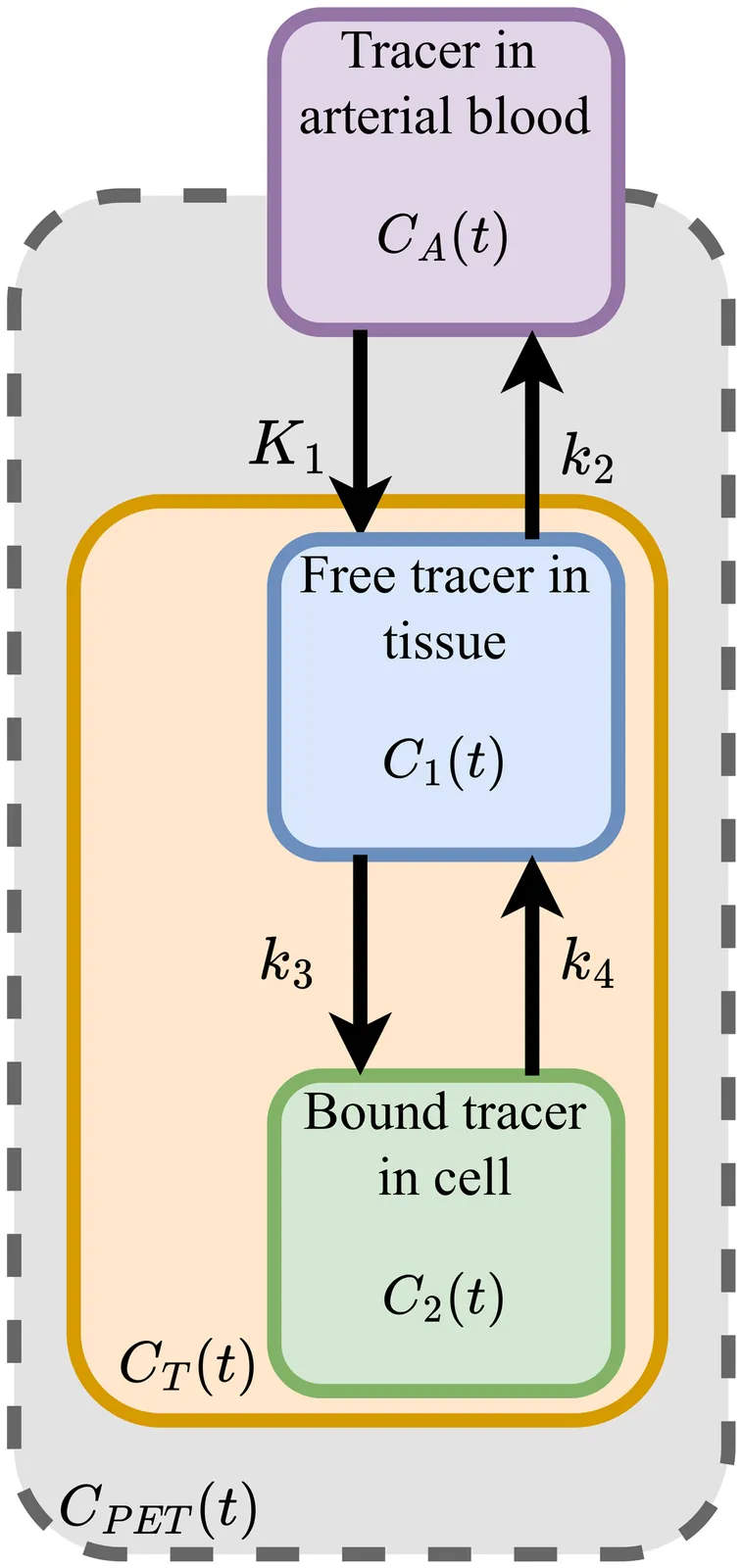
Physics-Informed Deep Learning for Improved Input Function Estimation in Motion-Blurred Dynamic [${}^{18}$F]FDG PET Images
Kinetic modeling enables \textit{in vivo} quantification of tracer uptake and glucose metabolism in [${}^{18}$F]Fluorodeoxyglucose ([${}^{18}$F]FDG) dynamic positron emission tomography (dPET) imaging of mice. However, kinetic modeling requires the accurate determination of the arterial input function (AIF) during imaging, which is time-consuming and invasive. Recent studies have shown the efficacy of using deep learning to directly predict the input function, surpassing established methods such as the image-derived input function (IDIF). In this work, we trained a physics-informed deep learning-based input function prediction model (PIDLIF) to estimate the AIF directly from the PET images, incorporating a kinetic modeling loss during training. The proposed method uses a two-tissue compartment model over two regions, the myocardium and brain of the mice, and is trained on a dataset of 70 [${}^{18}$F]FDG dPET images of mice accompanied by the measured AIF during imaging. The proposed method had comparable performance to the network without a physics-informed loss, and when sudden movement causing blurring in the images was simulated, the PIDLIF model maintained high performance in severe cases of image degradation. The proposed physics-informed method exhibits an improved robustness that is promoted by physically constraining the problem, enforcing consistency for out-of-distribution samples. In conclusion, the PIDLIF model offers insight into the effects of leveraging physiological distribution mechanics in mice to guide a deep learning-based AIF prediction network in images with severe degradation as a result of blurring due to movement during imaging.
2510.212811
Oct 2025Quantitative Methods
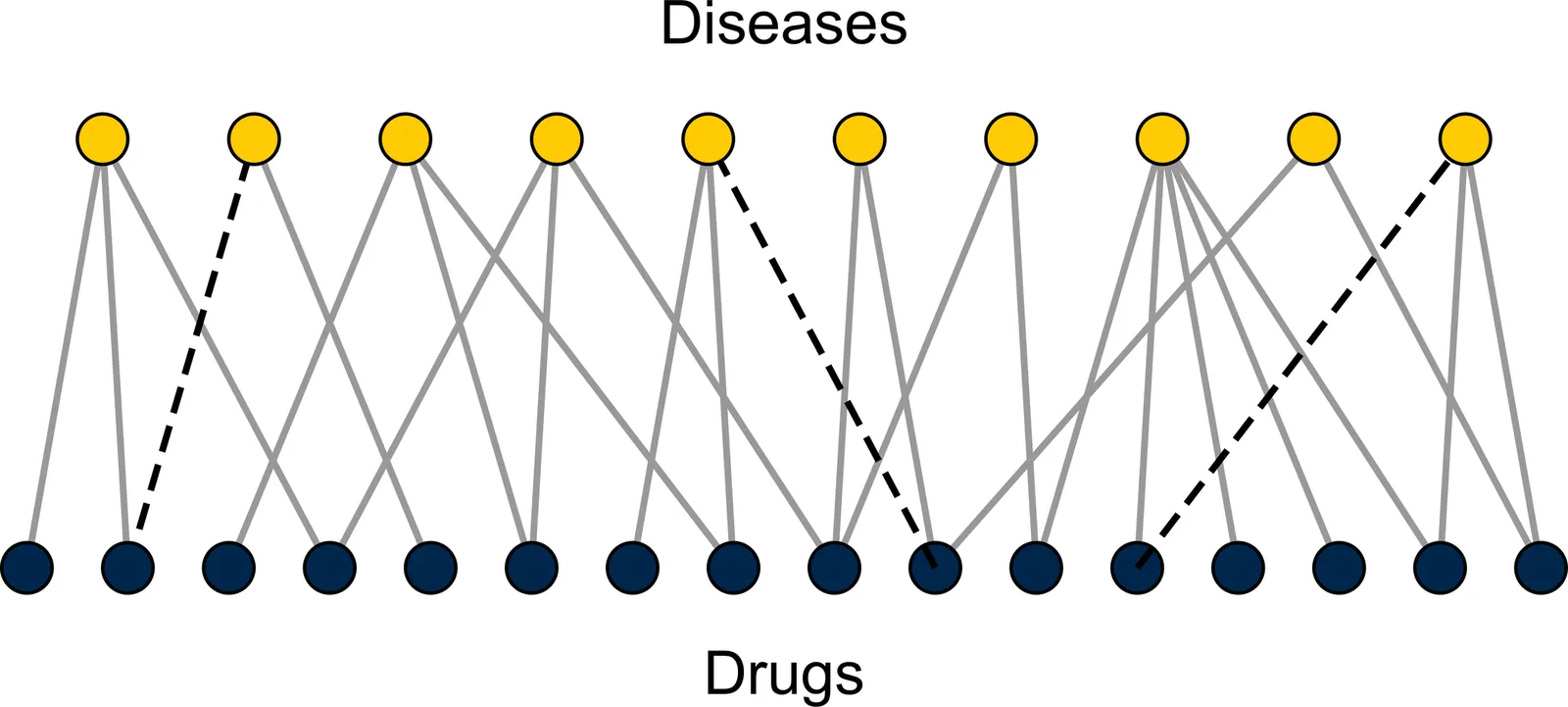
Drug-disease networks and drug repurposing
Repurposing existing drugs to treat new diseases is a cost-effective alternative to de novo drug development, but there are millions of potential drug-disease combinations to be considered with only a small fraction being viable. In silico predictions of drug-disease associations can be invaluable for reducing the size of the search space. In this work we present a novel network of drugs and the diseases they treat, compiled using a combination of existing textual and machine-readable databases, natural-language processing tools, and hand curation, and analyze it using network-based link prediction methods to identify potential drug-disease combinations. We measure the efficacy of these methods using cross-validation tests and find that several methods, particularly those based on graph embedding and network model fitting, achieve impressive prediction performance, significantly better than previous approaches, with area under the ROC curve above 0.95 and average precision almost a thousand times better than chance.
2510.199481
Oct 2025Quantitative Methods
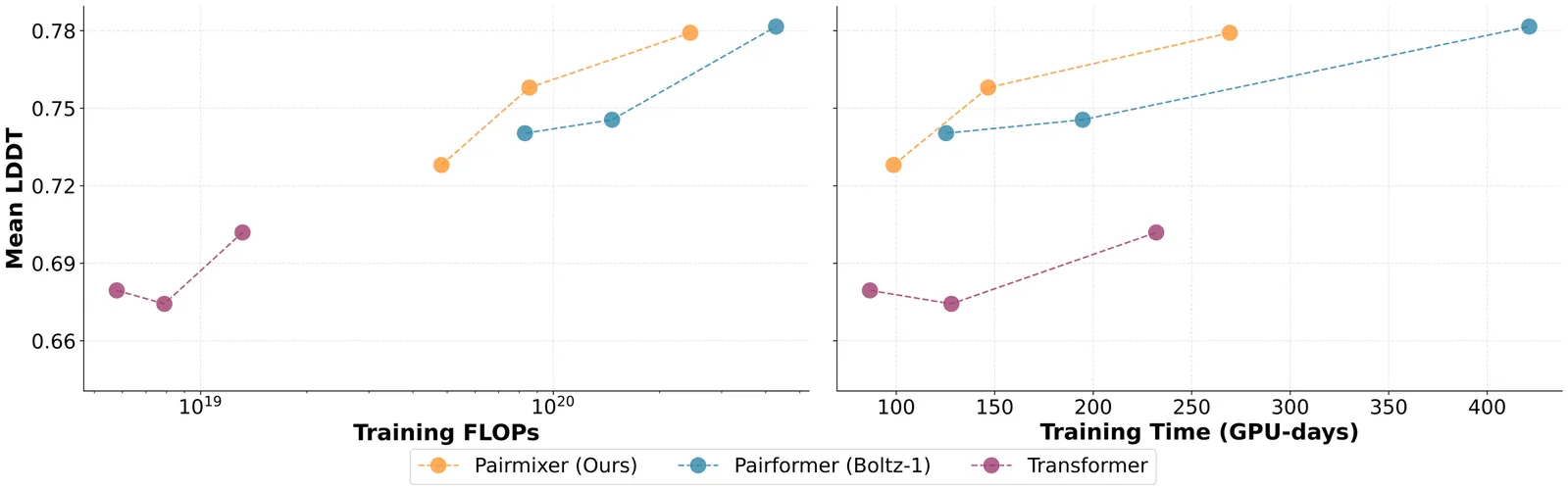
Triangle Multiplication Is All You Need For Biomolecular Structure Representations
AlphaFold has transformed protein structure prediction, but emerging applications such as virtual ligand screening, proteome-wide folding, and de novo binder design demand predictions at a massive scale, where runtime and memory costs become prohibitive. A major bottleneck lies in the Pairformer backbone of AlphaFold3-style models, which relies on computationally expensive triangular primitives-especially triangle attention-for pairwise reasoning. We introduce Pairmixer, a streamlined alternative that eliminates triangle attention while preserving higher-order geometric reasoning capabilities that are critical for structure prediction. Pairmixer substantially improves computational efficiency, matching state-of-the-art structure predictors across folding and docking benchmarks, delivering up to 4x faster inference on long sequences while reducing training cost by 34%. Its efficiency alleviates the computational burden of downstream applications such as modeling large protein complexes, high-throughput ligand and binder screening, and hallucination-based design. Within BoltzDesign, for example, Pairmixer delivers over 2x faster sampling and scales to sequences ~30% longer than the memory limits of Pairformer. Code is available at https://github.com/genesistherapeutics/pairmixer.
2510.188701
Oct 2025Quantitative Methods

Identifying multi-omics interactions for lung cancer drug targets discovery using Kernel Machine Regression
Cancer exhibits diverse and complex phenotypes driven by multifaceted molecular interactions. Recent biomedical research has emphasized the comprehensive study of such diseases by integrating multi-omics datasets (genome, proteome, transcriptome, epigenome). This approach provides an efficient method for identifying genetic variants associated with cancer and offers a deeper understanding of how the disease develops and spreads. However, it is challenging to comprehend complex interactions among the features of multi-omics datasets compared to single omics. In this paper, we analyze lung cancer multi-omics datasets from The Cancer Genome Atlas (TCGA). Using four statistical methods, LIMMA, the T test, Canonical Correlation Analysis (CCA), and the Wilcoxon test, we identified differentially expressed genes across gene expression, DNA methylation, and miRNA expression data. We then integrated these multi-omics data using the Kernel Machine Regression (KMR) approach. Our findings reveal significant interactions among the three omics: gene expression, miRNA expression, and DNA methylation in lung cancer. From our data analysis, we identified 38 genes significantly associated with lung cancer. From our data analysis, we identified 38 genes significantly associated with lung cancer. Among these, eight genes of highest ranking (PDGFRB, PDGFRA, SNAI1, ID1, FGF11, TNXB, ITGB1, ZIC1) were highlighted by rigorous statistical analysis. Furthermore, in silico studies identified three top-ranked potential candidate drugs (Selinexor, Orapred, and Capmatinib) that could play a crucial role in the treatment of lung cancer. These proposed drugs are also supported by the findings of other independent studies, which underscore their potential efficacy in the fight against lung cancer.
2510.160932
Oct 2025Genomics
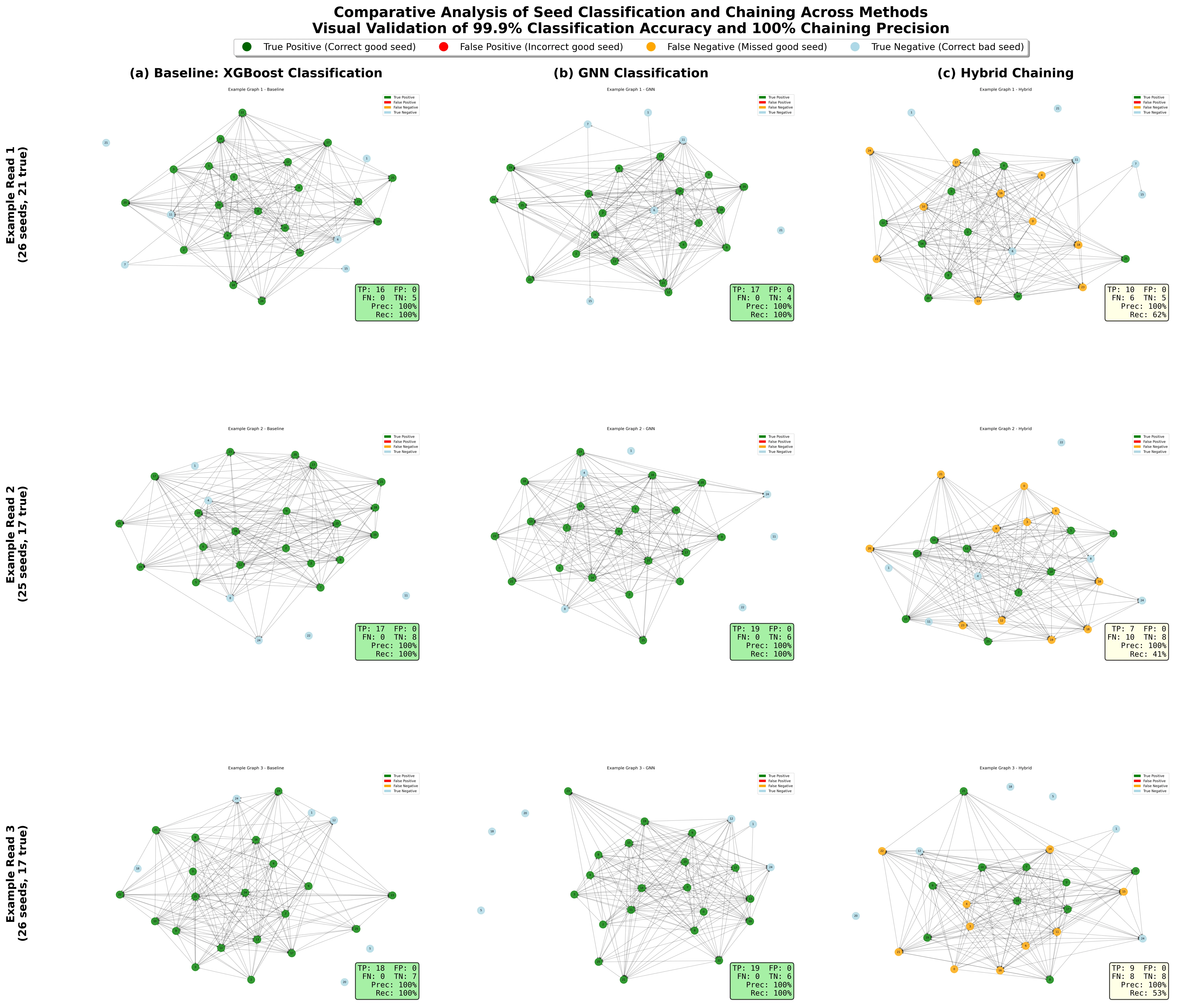
AGNES: Adaptive Graph Neural Network and Dynamic Programming Hybrid Framework for Real-Time Nanopore Seed Chaining
Nanopore sequencing enables real-time long-read DNA sequencing with reads exceeding 10 kilobases, but inherent error rates of 12-15 percent present significant computational challenges for read alignment. The critical seed chaining step must connect exact k-mer matches between reads and reference genomes while filtering spurious matches, yet state-of-the-art methods rely on fixed gap penalty functions unable to adapt to varying genomic contexts including tandem repeats and structural variants. This paper presents RawHash3, a hybrid framework combining graph neural networks with classical dynamic programming for adaptive seed chaining that maintains real-time performance while providing statistical guarantees. We formalize seed chaining as graph learning where seeds constitute nodes with 12-dimensional feature vectors and edges encode 8-dimensional spatial relationships including gap consistency. Our architecture employs three-layer EdgeConv GNN with confidence-based method selection that dynamically switches between learned guidance and algorithmic fallback. Comprehensive evaluation on 1,000 synthetic nanopore reads with 5,200 test seeds demonstrates RawHash3 achieves 99.94 percent precision and 40.07 percent recall, representing statistically significant 25.0 percent relative improvement over baseline with p less than 0.001. The system maintains median inference latency of 1.59ms meeting real-time constraints, while demonstrating superior robustness with 100 percent success rate under 20 percent label corruption versus baseline degradation to 30.3 percent. Cross-validation confirms stability establishing graph neural networks as viable approach for production genomics pipelines.
2510.160131
Oct 2025Genomics
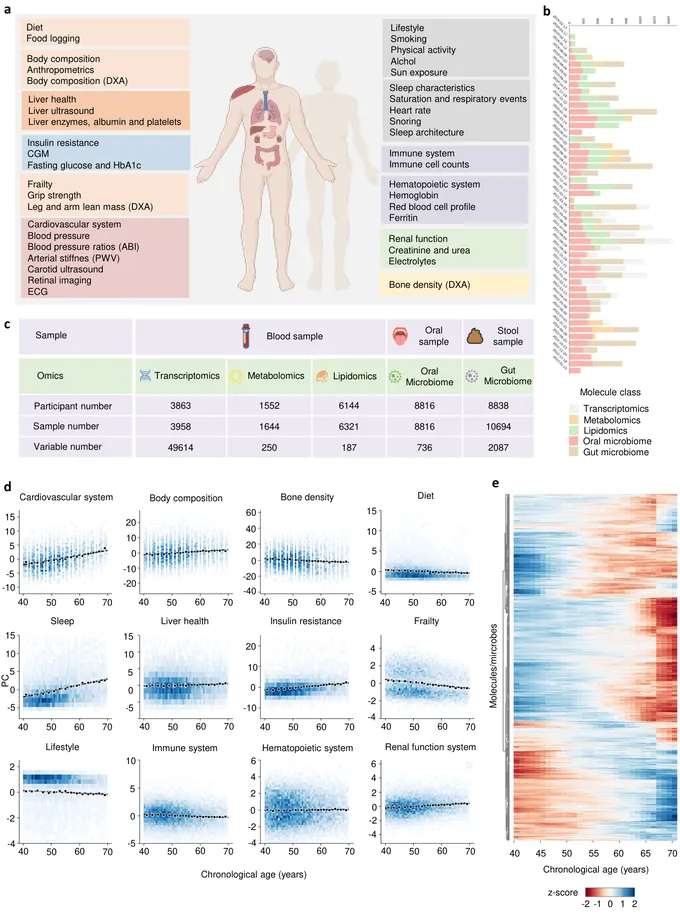
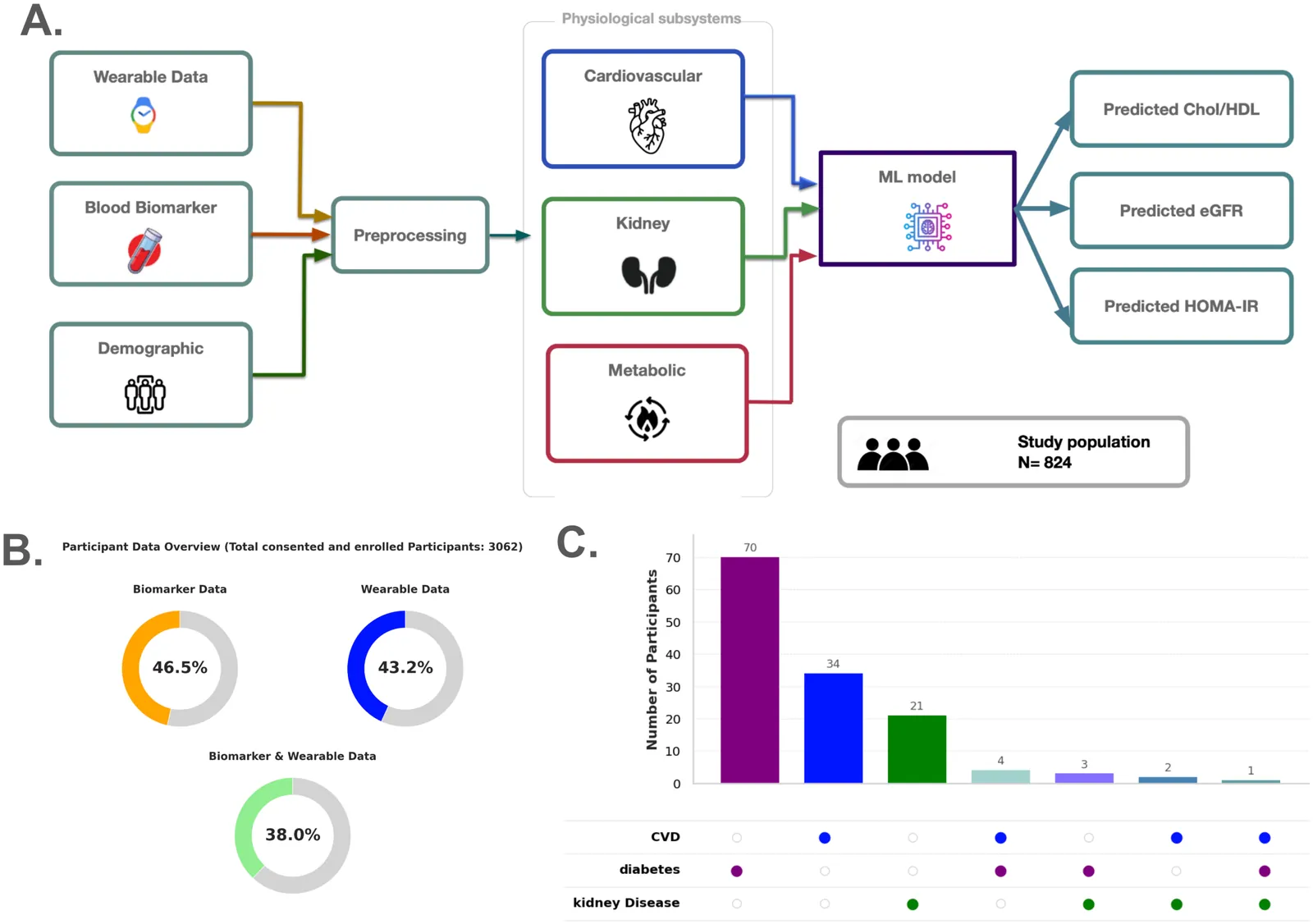
Phenome-Wide Multi-Omics Integration Uncovers Distinct Archetypes of Human Aging
Aging is a highly complex and heterogeneous process that progresses at different rates across individuals, making biological age (BA) a more accurate indicator of physiological decline than chronological age. While previous studies have built aging clocks using single-omics data, they often fail to capture the full molecular complexity of human aging. In this work, we leveraged the Human Phenotype Project, a large-scale cohort of 10,000 adults aged 40-70 years, with extensive longitudinal profiling that includes clinical, behavioral, environmental, and multi-omics datasets spanning transcriptomics, lipidomics, metabolomics, and the microbiome. By employing advanced machine learning frameworks capable of modeling nonlinear biological dynamics, we developed and rigorously validated a multi-omics aging clock that robustly predicts diverse health outcomes and future disease risk. Unsupervised clustering of the integrated molecular profiles from multi-omics uncovered distinct biological subtypes of aging, revealing striking heterogeneity in aging trajectories and pinpointing pathway-specific alterations associated with different aging patterns. These findings demonstrate the power of multi-omics integration to decode the molecular landscape of aging and lay the groundwork for personalized healthspan monitoring and precision strategies to prevent age-related diseases.
2510.123841
Oct 2025Genomics
PRISM: Enhancing Protein Inverse Folding through Fine-Grained Retrieval on Structure-Sequence Multimodal Representations
Designing protein sequences that fold into a target three-dimensional structure, known as the inverse folding problem, is central to protein engineering but remains challenging due to the vast sequence space and the importance of local structural constraints. Existing deep learning approaches achieve strong recovery rates, yet they lack explicit mechanisms to reuse fine-grained structure-sequence patterns that are conserved across natural proteins. We present PRISM, a multimodal retrieval-augmented generation framework for inverse folding that retrieves fine-grained representations of potential motifs from known proteins and integrates them with a hybrid self-cross attention decoder. PRISM is formulated as a latent-variable probabilistic model and implemented with an efficient approximation, combining theoretical grounding with practical scalability. Across five benchmarks (CATH-4.2, TS50, TS500, CAMEO 2022, and the PDB date split), PRISM establishes new state of the art in both perplexity and amino acid recovery, while also improving foldability metrics (RMSD, TM-score, pLDDT), demonstrating that fine-grained multimodal retrieval is a powerful and efficient paradigm for protein sequence design.
2510.117501
Oct 2025Quantitative Methods
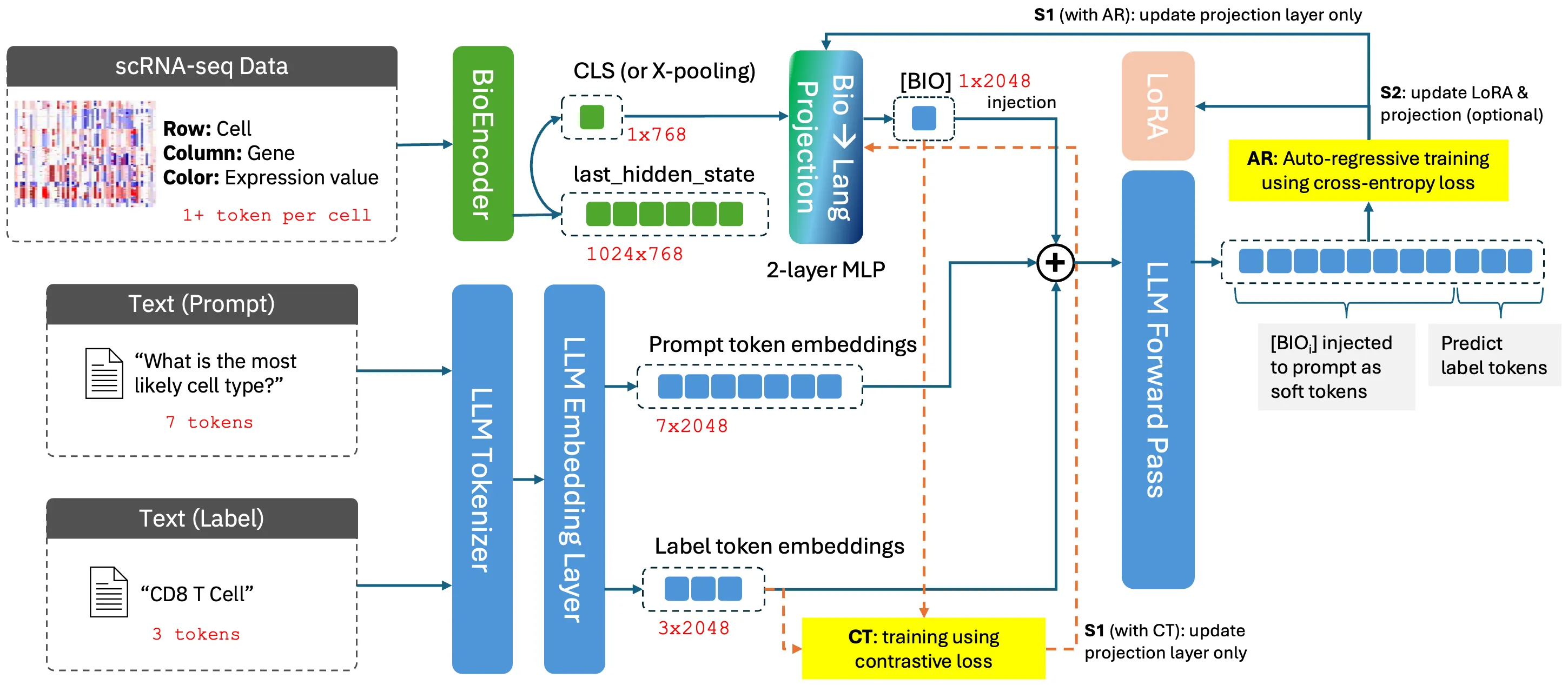
BioVERSE: Representation Alignment of Biomedical Modalities to LLMs for Multi-Modal Reasoning
Recent advances in large language models (LLMs) and biomedical foundation models (BioFMs) have achieved strong results in biological text reasoning, molecular modeling, and single-cell analysis, yet they remain siloed in disjoint embedding spaces, limiting cross-modal reasoning. We present BIOVERSE (Biomedical Vector Embedding Realignment for Semantic Engagement), a two-stage approach that adapts pretrained BioFMs as modality encoders and aligns them with LLMs through lightweight, modality-specific projection layers. The approach first aligns each modality to a shared LLM space through independently trained projections, allowing them to interoperate naturally, and then applies standard instruction tuning with multi-modal data to bring them together for downstream reasoning. By unifying raw biomedical data with knowledge embedded in LLMs, the approach enables zero-shot annotation, cross-modal question answering, and interactive, explainable dialogue. Across tasks spanning cell-type annotation, molecular description, and protein function reasoning, compact BIOVERSE configurations surpass larger LLM baselines while enabling richer, generative outputs than existing BioFMs, establishing a foundation for principled multi-modal biomedical reasoning.
2510.014281
Oct 2025Quantitative Methods
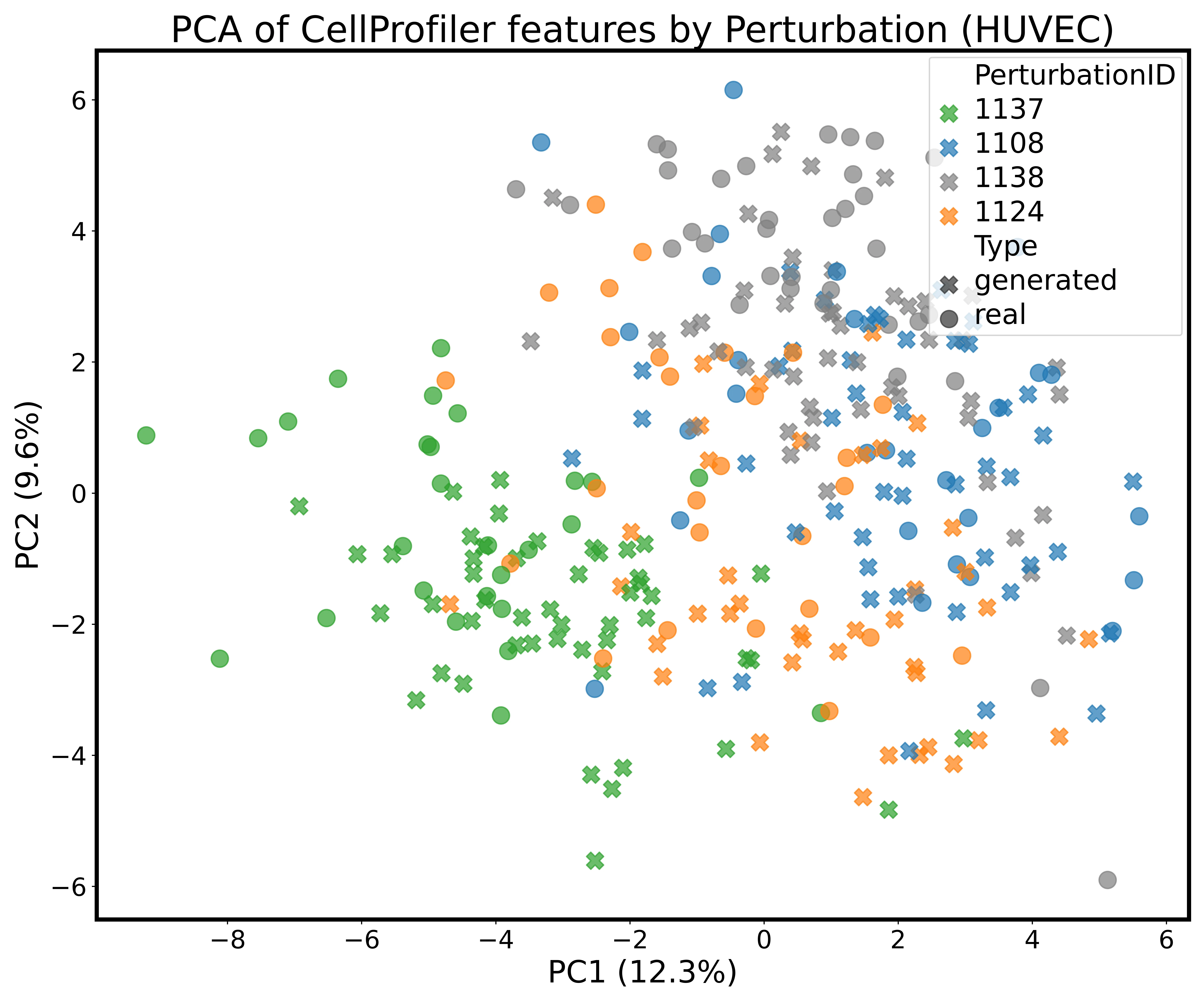
MorphGen: Controllable and Morphologically Plausible Generative Cell-Imaging
Simulating in silico cellular responses to interventions is a promising direction to accelerate high-content image-based assays, critical for advancing drug discovery and gene editing. To support this, we introduce MorphGen, a state-of-the-art diffusion-based generative model for fluorescent microscopy that enables controllable generation across multiple cell types and perturbations. To capture biologically meaningful patterns consistent with known cellular morphologies, MorphGen is trained with an alignment loss to match its representations to the phenotypic embeddings of OpenPhenom, a state-of-the-art biological foundation model. Unlike prior approaches that compress multichannel stains into RGB images -- thus sacrificing organelle-specific detail -- MorphGen generates the complete set of fluorescent channels jointly, preserving per-organelle structures and enabling a fine-grained morphological analysis that is essential for biological interpretation. We demonstrate biological consistency with real images via CellProfiler features, and MorphGen attains an FID score over 35% lower than the prior state-of-the-art MorphoDiff, which only generates RGB images for a single cell type. Code is available at https://github.com/czi-ai/MorphGen.
2510.012981
Oct 2025Quantitative Methods