Trending in Biomolecules
Generative AI for Enzyme Design and Biocatalysis
Sparked by innovations in generative artificial intelligence (AI), the field of protein design has undergone a paradigm shift with an explosion of new models for optimizing existing enzymes or creating them from scratch. After more than one decade of low success rates for computationally designed enzymes, generative AI models are now frequently used for designing proficient enzymes. Here, we provide a comprehensive overview and classification of generative AI models for enzyme design, highlighting models with experimental validation relevant to real-world settings and outlining their respective limitations. We argue that generative AI models now have the maturity to create and optimize enzymes for industrial applications. Wider adoption of generative AI models with experimental feedback loops can speed up the development of biocatalysts and serve as a community assessment to inform the next generation of models.
2602.03779
Feb 2026Biomolecules
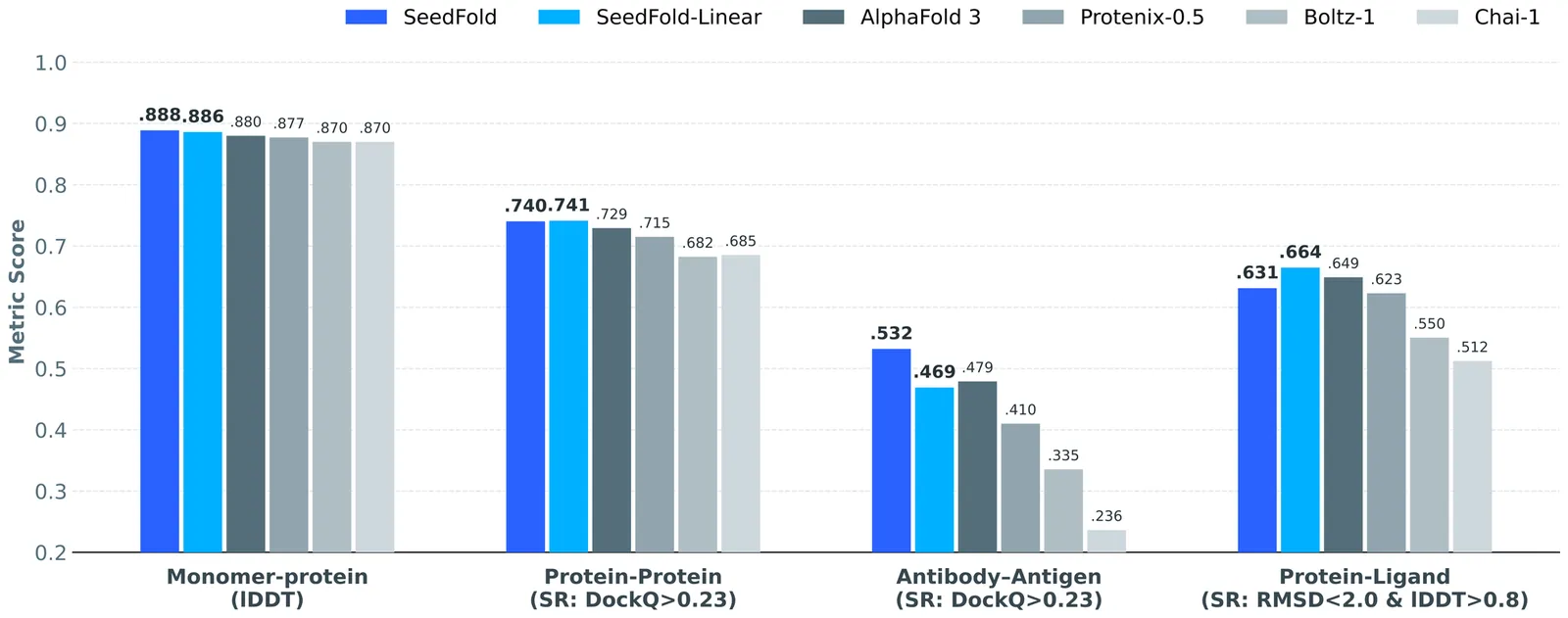
SeedFold: Scaling Biomolecular Structure Prediction
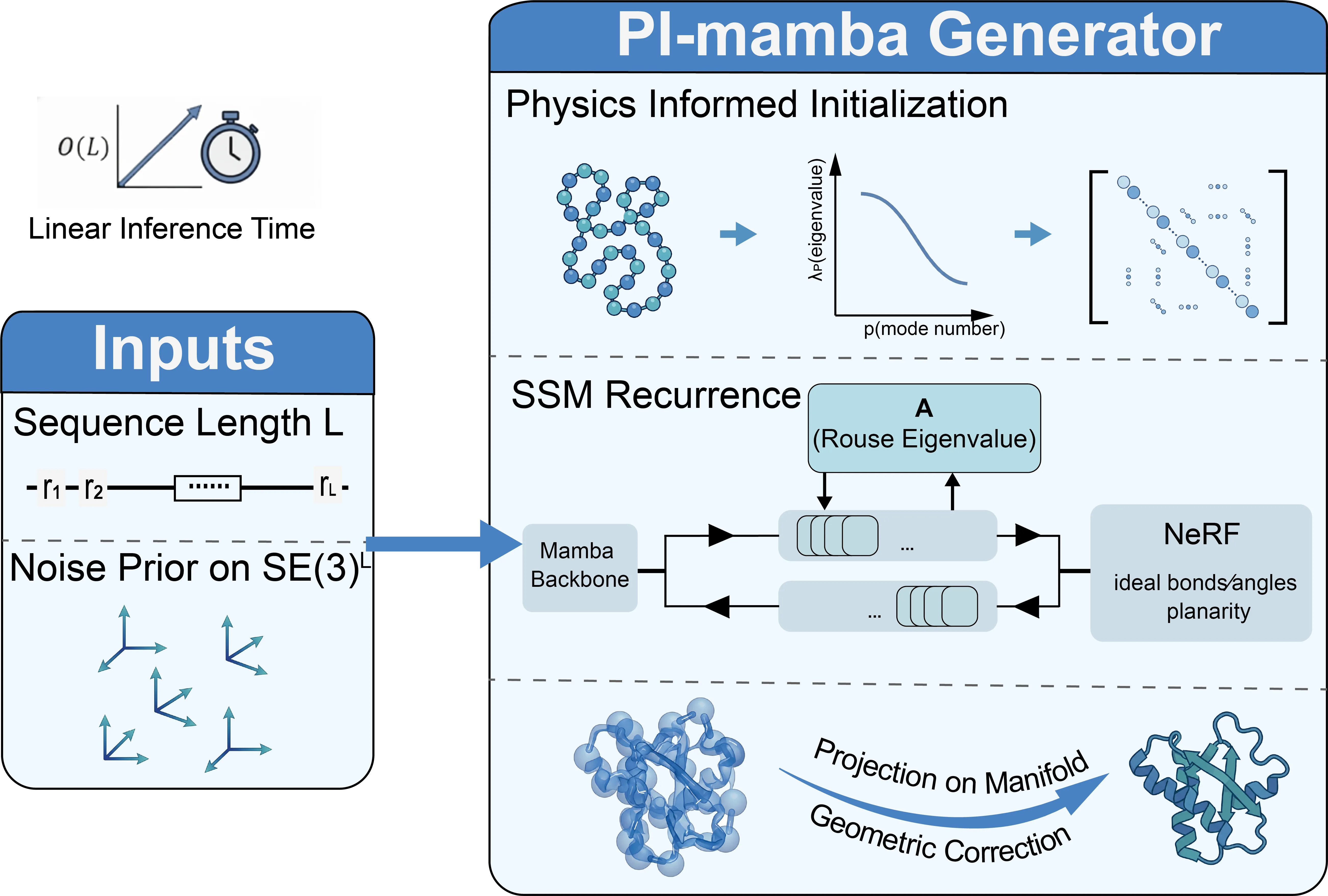
Highly accurate biomolecular structure prediction is a key component of developing biomolecular foundation models, and one of the most critical aspects of building foundation models is identifying the recipes for scaling the model. In this work, we present SeedFold, a folding model that successfully scales up the model capacity. Our contributions are threefold: first, we identify an effective width-scaling strategy for the Pairformer to increase representation capacity; second, we introduce a novel linear triangular attention that reduces computational complexity to enable efficient scaling; finally, we construct a large-scale distillation dataset to substantially enlarge the training set. Experiments on FoldBench show that SeedFold outperforms AlphaFold3 on most protein-related tasks.
2512.243541
Dec 2025Biomolecules
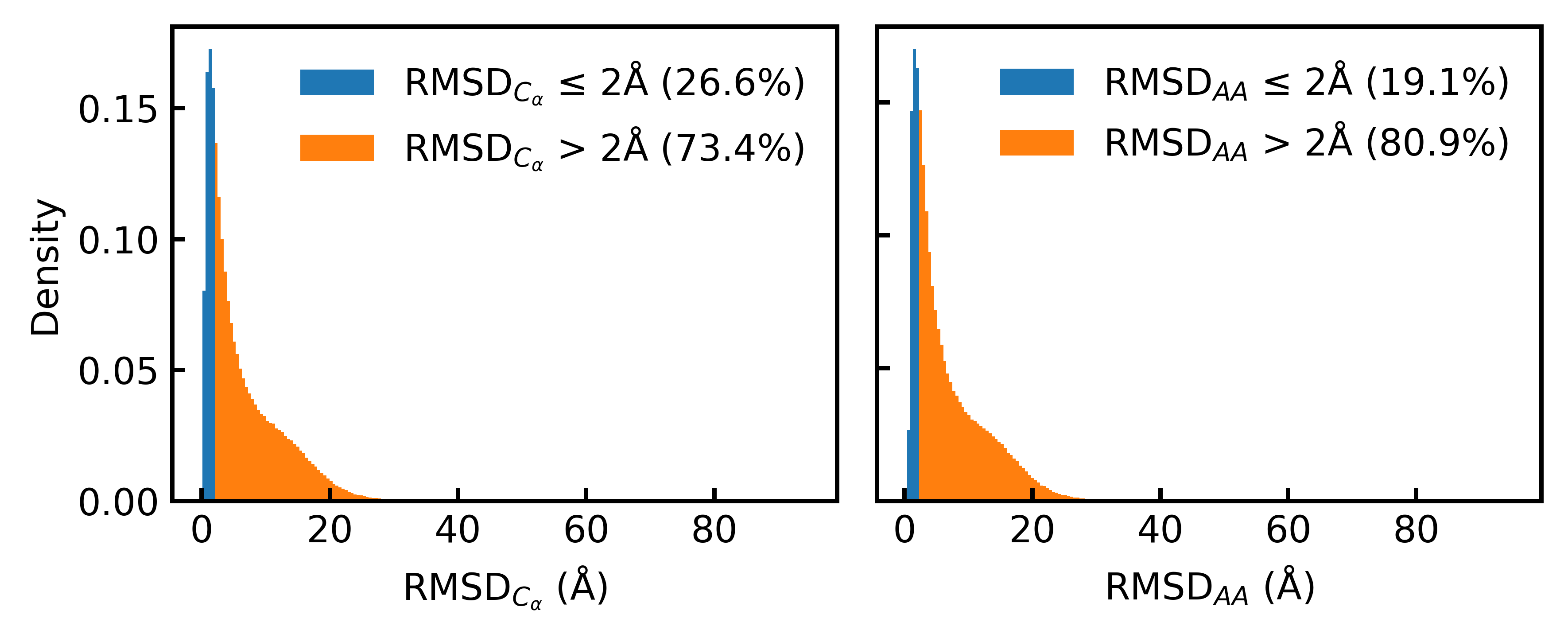
Consistent Synthetic Sequences Unlock Structural Diversity in Fully Atomistic De Novo Protein Design
High-quality training datasets are crucial for the development of effective protein design models, but existing synthetic datasets often include unfavorable sequence-structure pairs, impairing generative model performance. We leverage ProteinMPNN, whose sequences are experimentally favorable as well as amenable to folding, together with structure prediction models to align high-quality synthetic structures with recoverable synthetic sequences. In that way, we create a new dataset designed specifically for training expressive, fully atomistic protein generators. By retraining La-Proteina, which models discrete residue type and side chain structure in a continuous latent space, on this dataset, we achieve new state-of-the-art results, with improvements of +54% in structural diversity and +27% in co-designability. To validate the broad utility of our approach, we further introduce Proteina Atomistica, a unified flow-based framework that jointly learns the distribution of protein backbone structure, discrete sequences, and atomistic side chains without latent variables. We again find that training on our new sequence-structure data dramatically boosts benchmark performance, improving \method's structural diversity by +73% and co-designability by +5%. Our work highlights the critical importance of aligned sequence-structure data for training high-performance de novo protein design models. All data will be publicly released.
2512.01976
Dec 2025Biomolecules