Other Quantitative Biology
arXiv:q-bio.OT
Quantitative biology not fitting other q-bio categories.
Quantitative biology not fitting other q-bio categories.
MixRx uses Large Language Models (LLMs) to classify drug combination interactions as Additive, Synergistic, or Antagonistic, given a multi-drug patient history. We evaluate the performance of 4 models, GPT-2, Mistral Instruct 2.0, and the fine-tuned counterparts. Our results showed a potential for such an application, with the Mistral Instruct 2.0 Fine-Tuned model providing an average accuracy score on standard and perturbed datasets of 81.5%. This paper aims to further develop an upcoming area of research that evaluates if LLMs can be used for biological prediction tasks.
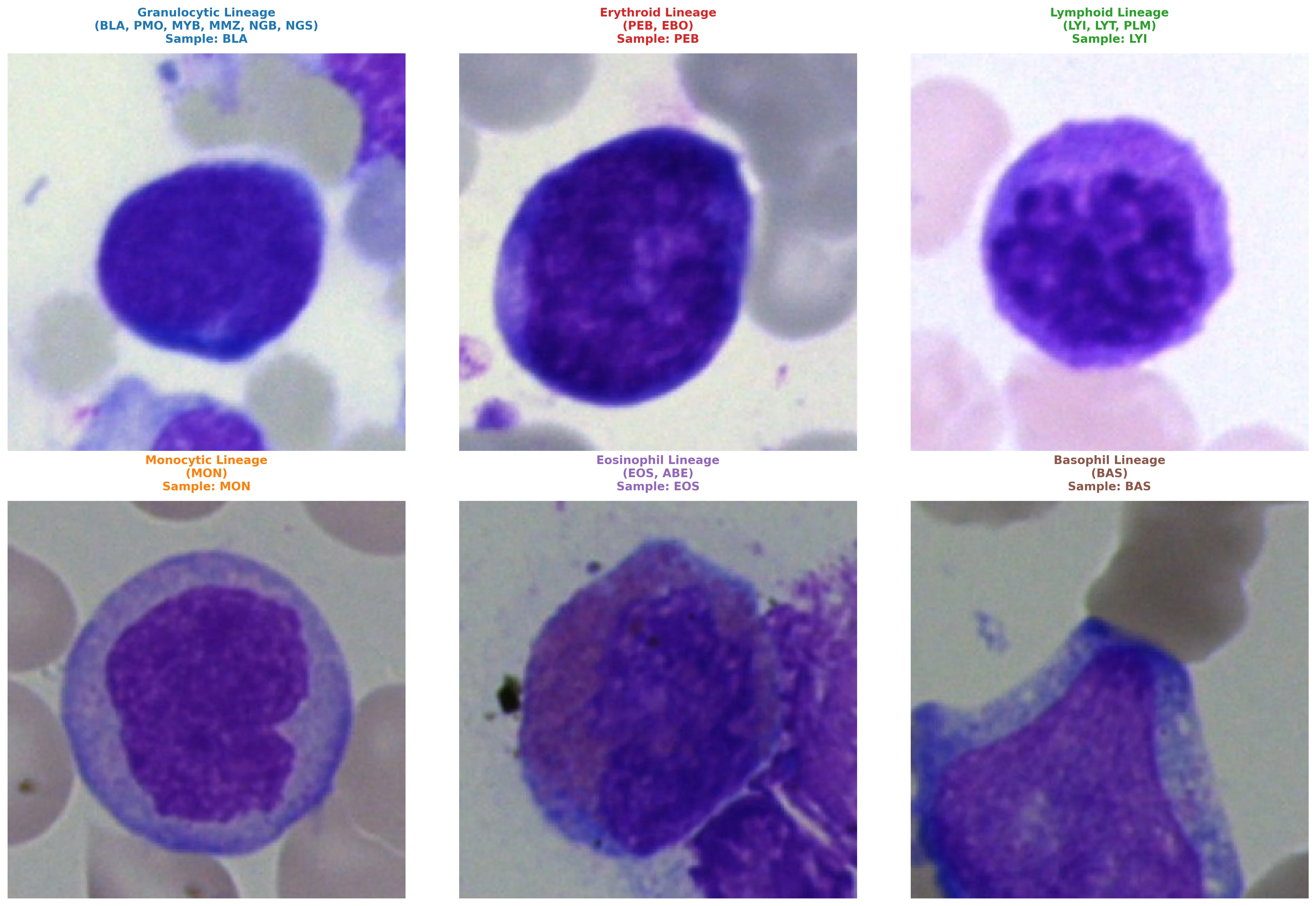
Bone marrow cell cytomorphology analysis is critical for the diagnosis of hematological malignancies but remains a labor-intensive process subject to significant inter-observer variability. While recent foundation models have shown promise in computational pathology, they often require extensive computational resources and fail to account for the asymmetric risks associated with clinical misdiagnosis. We introduce CytoDINO, a framework that achieves state-of-the-art performance on the Munich Leukemia Laboratory (MLL) dataset by fine-tuning DINOv3 using Low-Rank Adaptation (LoRA). Our primary contribution is a novel Hierarchical Focal Loss with Critical Penalties, which encodes biological relationships between cell lineages and explicitly penalizes clinically dangerous misclassifications (e.g., classifying blasts as normal cells). CytoDINO achieves an 88.2% weighted F1 score and 76.5% macro F1 on a held-out test set of 21 cell classes. By utilizing parameter-efficient fine-tuning with only 8% trainable parameters on a single NVIDIA RTX 5080, we demonstrate that consumer-grade hardware can match specialized infrastructure. Furthermore, confidence-based selective prediction yields 99.5% accuracy on 67% of samples, suggesting a viable pathway for clinical deployment where high-uncertainty cases are flagged for expert review
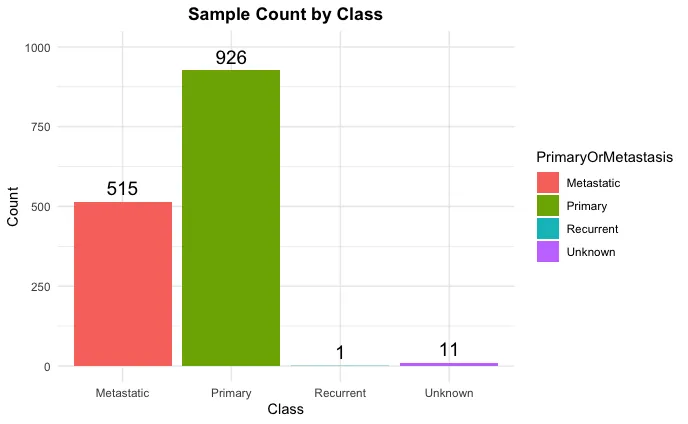
Metastasis is the leading cause of cancer-related mortality, yet most predictive models rely on shallow architectures and neglect patient-specific regulatory mechanisms. Here, we integrate classical machine learning and deep learning to predict metastatic potential across multiple cancer types. Gene expression profiles from the Cancer Cell Line Encyclopedia were combined with a transcription factor-target prior from DoRothEA, focusing on nine metastasis-associated regulators. After selecting differential genes using the Kruskal-Wallis test, ElasticNet, Random Forest, and XGBoost models were trained for benchmarking. Personalized gene regulatory networks were then constructed using PANDA and LIONESS and analyzed through a graph attention neural network (GATv2) to learn topological and expression-based representations. While XGBoost achieved the highest AUROC (0.7051), the GNN captured non-linear regulatory dependencies at the patient level. These results demonstrate that combining traditional machine learning with graph-based deep learning enables a scalable and interpretable framework for metastasis risk prediction in precision oncology.
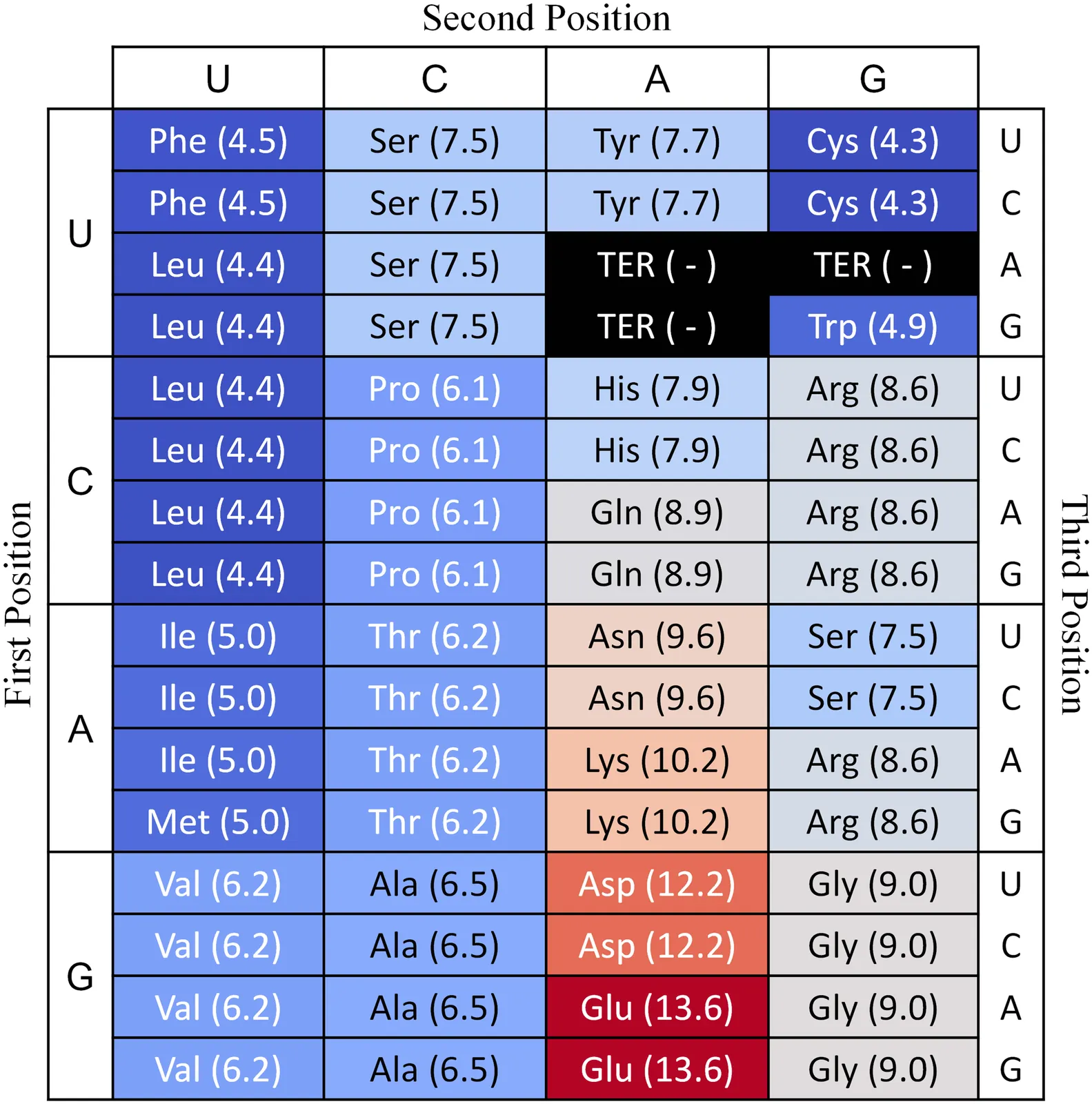
The origin and organizing principles of the genetic code remain fundamental puzzles in life science. The vanishingly low probability of the natural codon-to-amino acid mapping arising by chance has spurred the hypothesis that its structure is a solution optimized for robustness against mutations and translational errors. For the construction of effective molecular machines, the dictionary of encoded amino acids must also be diverse enough in physicochemical features. Here, we examine whether the standard genetic code can be understood as a near-optimal solution balancing these two objectives: minimizing error load and aligning codon assignments with the naturally occurring amino acid composition. Using simulated annealing, we explore this trade-off across a broad range of parameters. We find that the standard genetic code lies near local optima within the multidimensional parameter space. It is a highly effective solution that balances fidelity against resource availability constraints. These results suggest that the present genetic code reflects coevolution under conflicting pressures of fidelity and diversity, offering new insight into its emergence and evolution.

This paper details an observation that for more primitive organisms, such as some yeasts, the statistical distribution of the origins of replication sometimes looks remarkably like the distribution of eigenvalues from the Circular Orthogonal Ensemble (COE) of random matrices. This does not hold for more complex organisms, but a uniform thinning of the COE eigenvalues (which interpolates between the COE and uncorrelated, Poisson statistics) gives a platform to investigate characteristics of replication origin distribution in other species where data is available.

DNA has emerged as a promising alternative for long-term data storage due to its high capacity, durability, and low-energy potential. However, storing data in DNA presents several challenges. First, it requires complex and costly biochemical processes, making efficient compression crucial to reducing DNA synthesis time and cost. Second, these processes are prone to errors that must be avoided and/or corrected. In particular, homopolymers (repetitions of the same nucleotide) are a wellknown source of errors during the sequencing step. Avoiding such repetitions helps mitigate errors but introduces a constraint that may increase the data compression rate. In this paper, we propose two transcoding methods that address these two key challenges: reducing data rate and minimizing errors. The first method strictly enforces the error-minimization constraint by eliminating homopolymers of a certain length, at the cost of an increased data rate. In contrast, the second method accepts a slight increase in homopolymers. However, we show that these increases remain limited (2.14% increase in compression rate for the first method and 0.39% homopolymer rate for the second). These two approaches demonstrate that it is possible to efficiently constrain transcoding while balancing error minimization and compression performance.
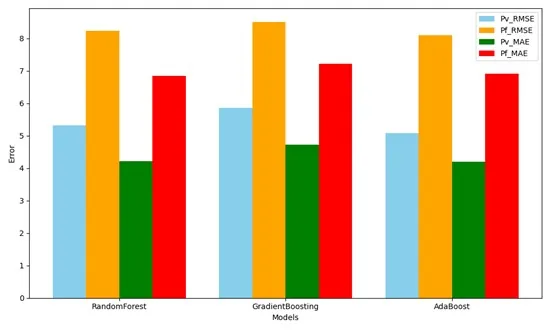
Malaria remains a major public health concern in Ethiopia, particularly in the Amhara Region, where seasonal and unpredictable transmission patterns make prevention and control challenging. Accurately forecasting malaria outbreaks is essential for effective resource allocation and timely interventions. This study proposes a hybrid predictive modeling framework that combines time-series forecasting, multi-output regression, and conventional regression-based prediction to forecast the incidence of malaria. Environmental variables, past malaria case data, and demographic information from Amhara Region health centers were used to train and validate the models. The multi-output regression approach enables the simultaneous prediction of multiple outcomes, including Plasmodium species-specific cases, temporal trends, and spatial variations, whereas the hybrid framework captures both seasonal patterns and correlations among predictors. The proposed model exhibits higher prediction accuracy than single-method approaches, exposing hidden patterns and providing valuable information to public health authorities. This study provides a valid and repeatable malaria incidence prediction framework that can support evidence-based decision-making, targeted interventions, and resource optimization in endemic areas.

Many people suffer from mental health problems but not everyone seeks professional help or has access to mental health care. AI chatbots have increasingly become a go-to for individuals who either have mental disorders or simply want someone to talk to. This paper presents a study on participants who have previously used chatbots and a scenario-based testing of large language model (LLM) chatbots. Our findings indicate that AI chatbots were primarily utilized as a "Five minute therapist" or as a non-judgmental companion. Participants appreciated the anonymity and lack of judgment from chatbots. However, there were concerns about privacy and the security of sensitive information. The scenario-based testing of LLM chatbots highlighted additional issues. Some chatbots were consistently reassuring, used emojis and names to add a personal touch, and were quick to suggest seeking professional help. However, there were limitations such as inconsistent tone, occasional inappropriate responses (e.g., casual or romantic), and a lack of crisis sensitivity, particularly in recognizing red flag language and escalating responses appropriately. These findings can inform both the technology and mental health care industries on how to better utilize AI chatbots to support individuals during challenging emotional periods.
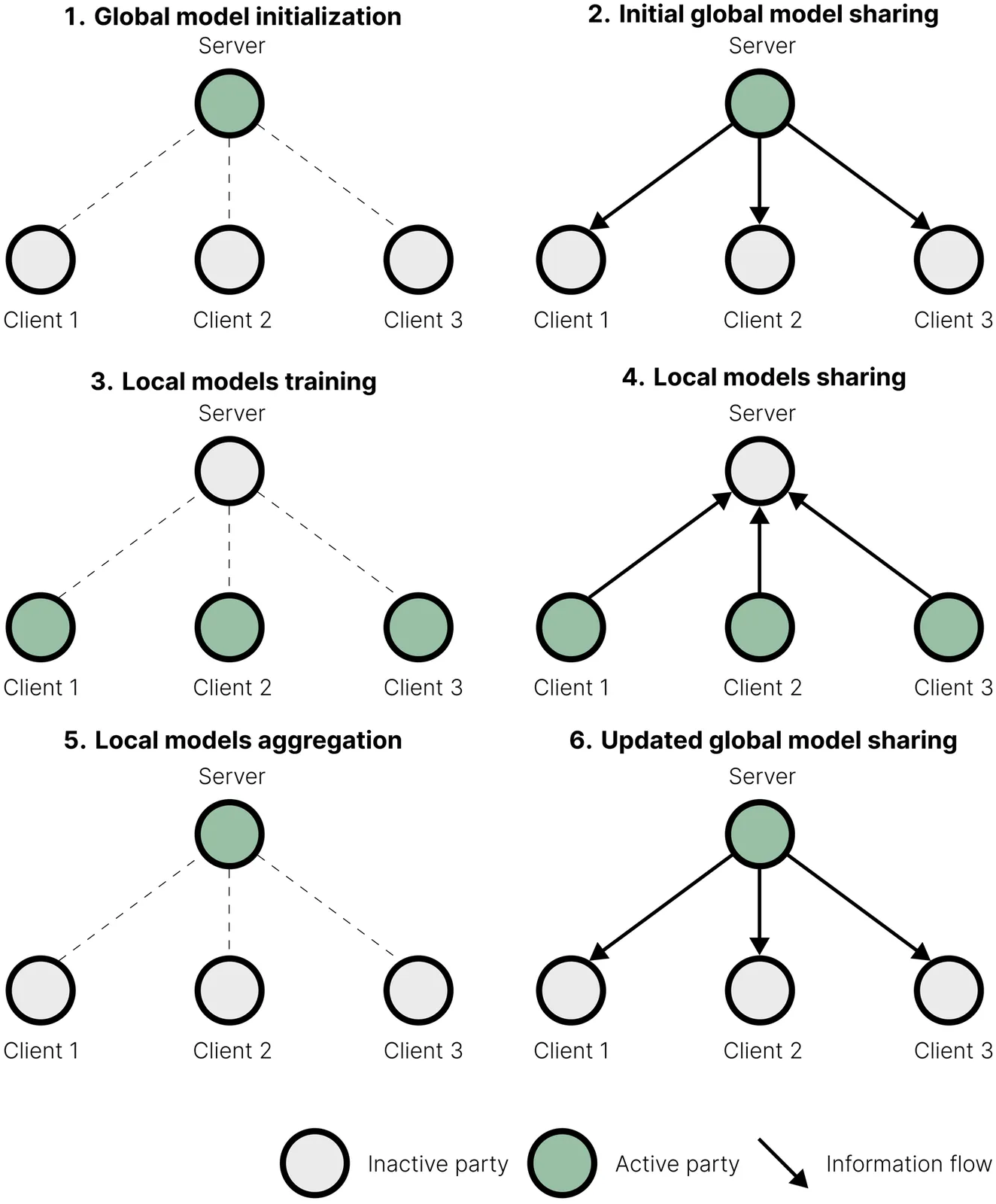
Federated learning leverages data across institutions to improve clinical discovery while complying with data-sharing restrictions and protecting patient privacy. This paper provides a gentle introduction to this approach in bioinformatics, and is the first to review key applications in proteomics, genome-wide association studies (GWAS), single-cell and multi-omics studies in their legal as well as methodological and infrastructural challenges. As the evolution of biobanks in genetics and systems biology has proved, accessing more extensive and varied data pools leads to a faster and more robust exploration and translation of results. More widespread use of federated learning may have a similar impact in bioinformatics, allowing academic and clinical institutions to access many combinations of genotypic, phenotypic and environmental information that are undercovered or not included in existing biobanks.
The translation of AI-generated brain metastases (BM) segmentation into clinical practice relies heavily on diverse, high-quality annotated medical imaging datasets. The BraTS-METS 2023 challenge has gained momentum for testing and benchmarking algorithms using rigorously annotated internationally compiled real-world datasets. This study presents the results of the segmentation challenge and characterizes the challenging cases that impacted the performance of the winning algorithms. Untreated brain metastases on standard anatomic MRI sequences (T1, T2, FLAIR, T1PG) from eight contributed international datasets were annotated in stepwise method: published UNET algorithms, student, neuroradiologist, final approver neuroradiologist. Segmentations were ranked based on lesion-wise Dice and Hausdorff distance (HD95) scores. False positives (FP) and false negatives (FN) were rigorously penalized, receiving a score of 0 for Dice and a fixed penalty of 374 for HD95. Eight datasets comprising 1303 studies were annotated, with 402 studies (3076 lesions) released on Synapse as publicly available datasets to challenge competitors. Additionally, 31 studies (139 lesions) were held out for validation, and 59 studies (218 lesions) were used for testing. Segmentation accuracy was measured as rank across subjects, with the winning team achieving a LesionWise mean score of 7.9. Common errors among the leading teams included false negatives for small lesions and misregistration of masks in space.The BraTS-METS 2023 challenge successfully curated well-annotated, diverse datasets and identified common errors, facilitating the translation of BM segmentation across varied clinical environments and providing personalized volumetric reports to patients undergoing BM treatment.
Many people suffer from mental health problems but not everyone seeks professional help or has access to mental health care. AI chatbots have increasingly become a go-to for individuals who either have mental disorders or simply want someone to talk to. This paper presents a study on participants who have previously used chatbots and a scenario-based testing of large language model (LLM) chatbots. Our findings indicate that AI chatbots were primarily utilized as a "Five minute therapist" or as a non-judgmental companion. Participants appreciated the anonymity and lack of judgment from chatbots. However, there were concerns about privacy and the security of sensitive information. The scenario-based testing of LLM chatbots highlighted additional issues. Some chatbots were consistently reassuring, used emojis and names to add a personal touch, and were quick to suggest seeking professional help. However, there were limitations such as inconsistent tone, occasional inappropriate responses (e.g., casual or romantic), and a lack of crisis sensitivity, particularly in recognizing red flag language and escalating responses appropriately. These findings can inform both the technology and mental health care industries on how to better utilize AI chatbots to support individuals during challenging emotional periods.
Federated learning leverages data across institutions to improve clinical discovery while complying with data-sharing restrictions and protecting patient privacy. This paper provides a gentle introduction to this approach in bioinformatics, and is the first to review key applications in proteomics, genome-wide association studies (GWAS), single-cell and multi-omics studies in their legal as well as methodological and infrastructural challenges. As the evolution of biobanks in genetics and systems biology has proved, accessing more extensive and varied data pools leads to a faster and more robust exploration and translation of results. More widespread use of federated learning may have a similar impact in bioinformatics, allowing academic and clinical institutions to access many combinations of genotypic, phenotypic and environmental information that are undercovered or not included in existing biobanks.
The translation of AI-generated brain metastases (BM) segmentation into clinical practice relies heavily on diverse, high-quality annotated medical imaging datasets. The BraTS-METS 2023 challenge has gained momentum for testing and benchmarking algorithms using rigorously annotated internationally compiled real-world datasets. This study presents the results of the segmentation challenge and characterizes the challenging cases that impacted the performance of the winning algorithms. Untreated brain metastases on standard anatomic MRI sequences (T1, T2, FLAIR, T1PG) from eight contributed international datasets were annotated in stepwise method: published UNET algorithms, student, neuroradiologist, final approver neuroradiologist. Segmentations were ranked based on lesion-wise Dice and Hausdorff distance (HD95) scores. False positives (FP) and false negatives (FN) were rigorously penalized, receiving a score of 0 for Dice and a fixed penalty of 374 for HD95. Eight datasets comprising 1303 studies were annotated, with 402 studies (3076 lesions) released on Synapse as publicly available datasets to challenge competitors. Additionally, 31 studies (139 lesions) were held out for validation, and 59 studies (218 lesions) were used for testing. Segmentation accuracy was measured as rank across subjects, with the winning team achieving a LesionWise mean score of 7.9. Common errors among the leading teams included false negatives for small lesions and misregistration of masks in space.The BraTS-METS 2023 challenge successfully curated well-annotated, diverse datasets and identified common errors, facilitating the translation of BM segmentation across varied clinical environments and providing personalized volumetric reports to patients undergoing BM treatment.