Trending in Materials Science
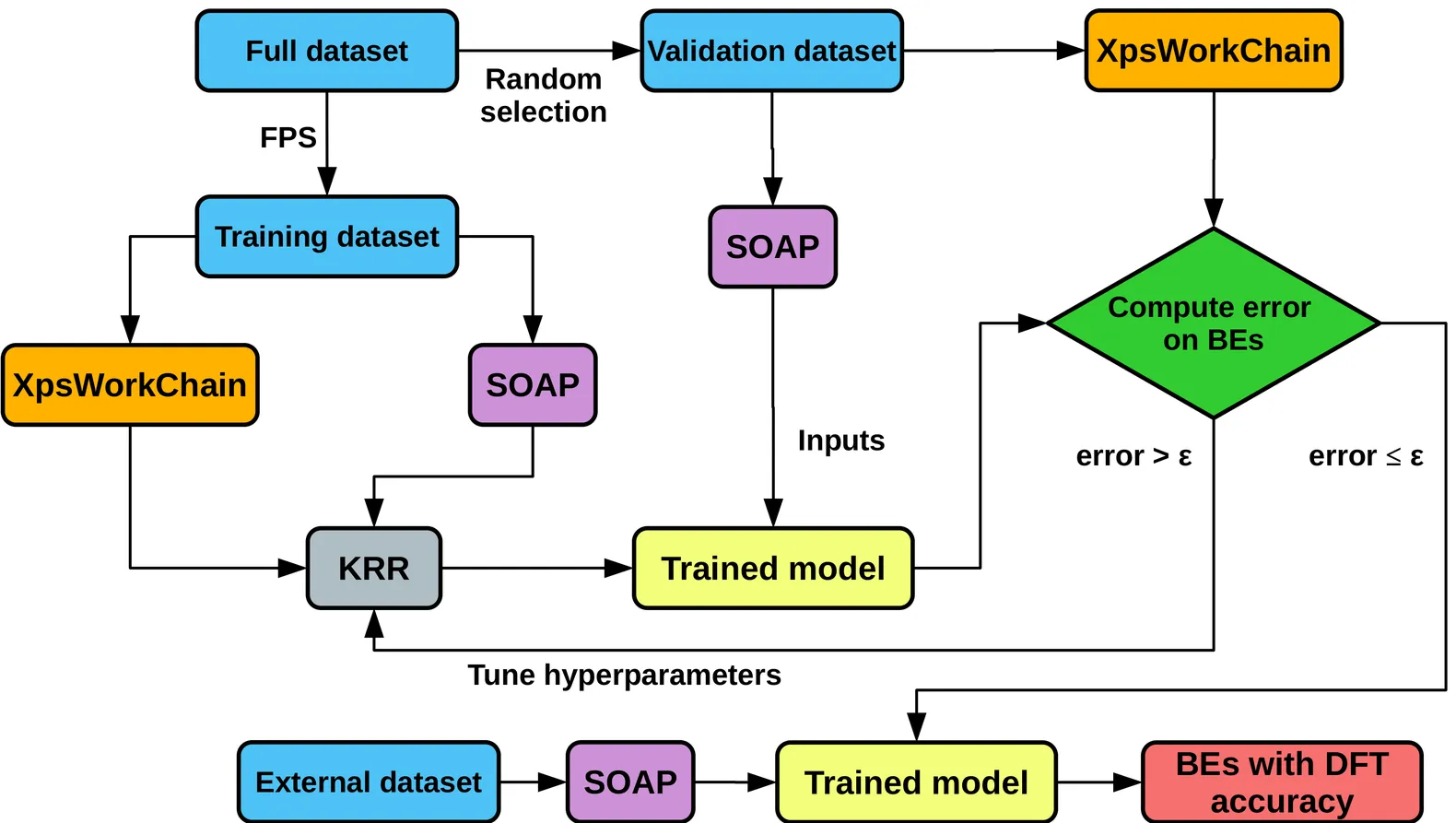
Tracking the Lithiation State of Li$_x$Si from Machine-Learned XPS Binding Energies
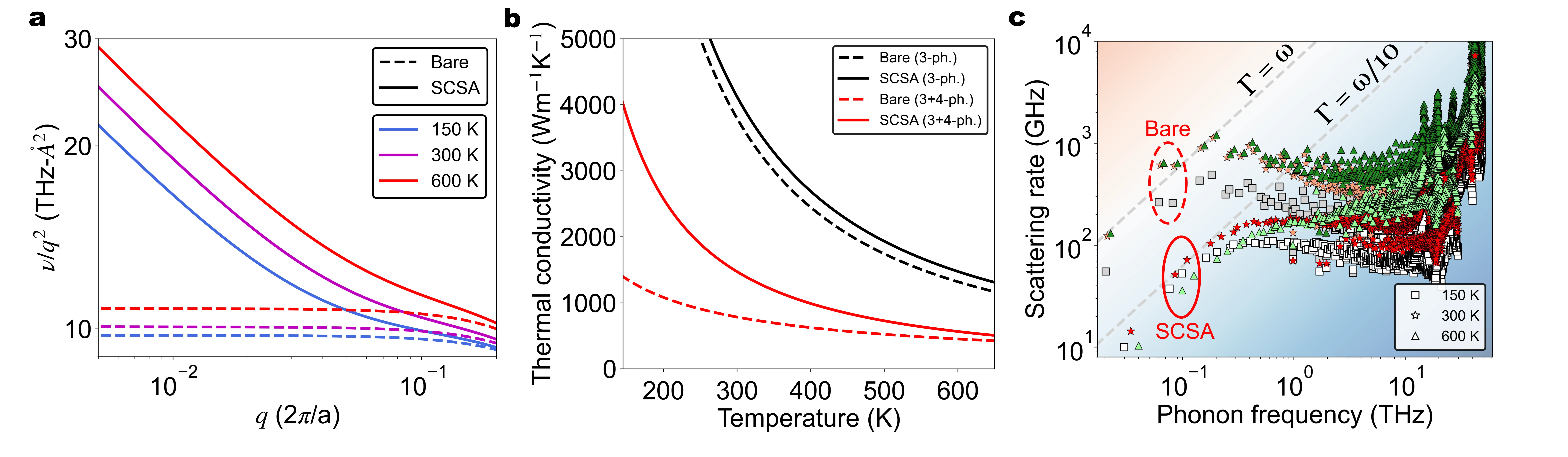
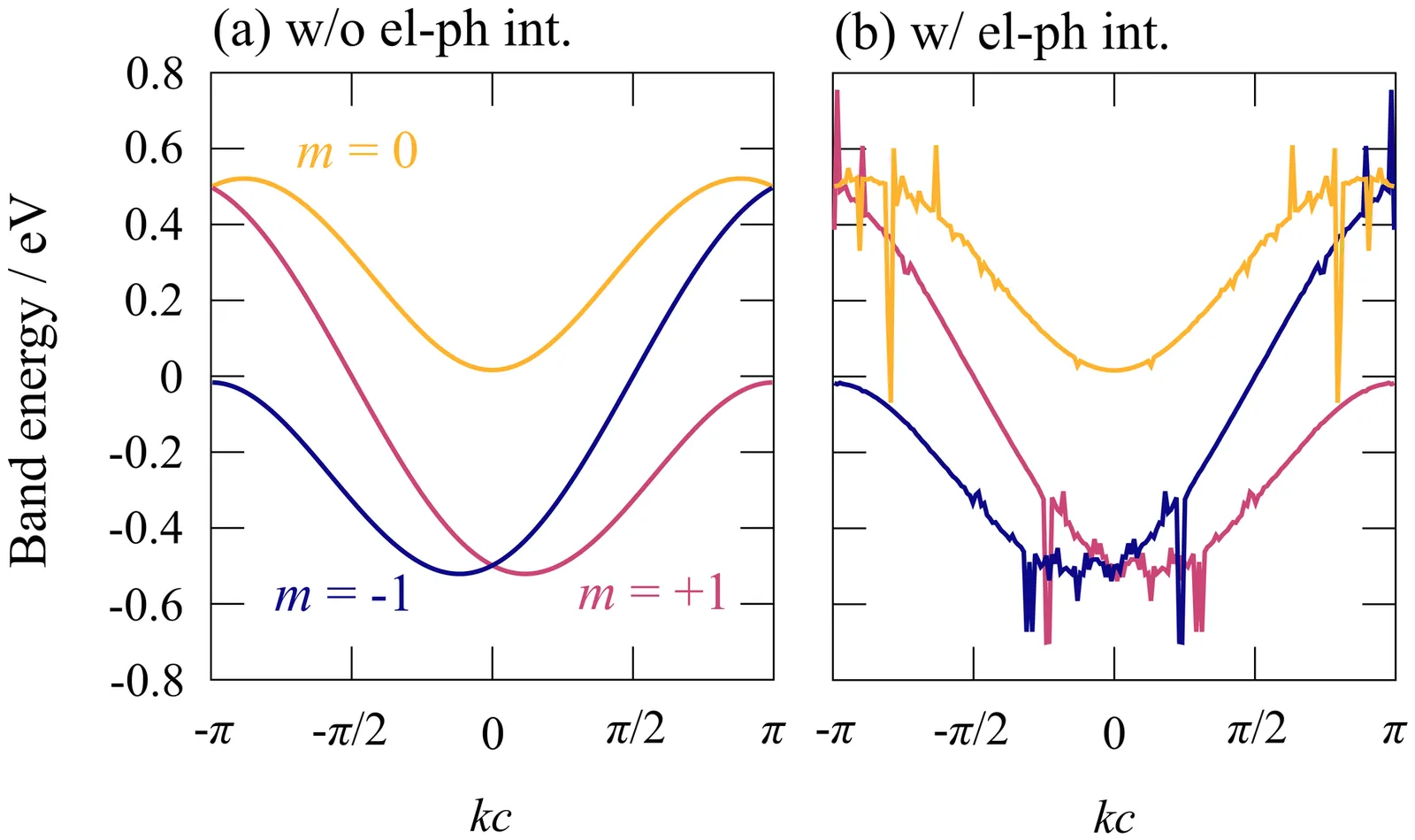
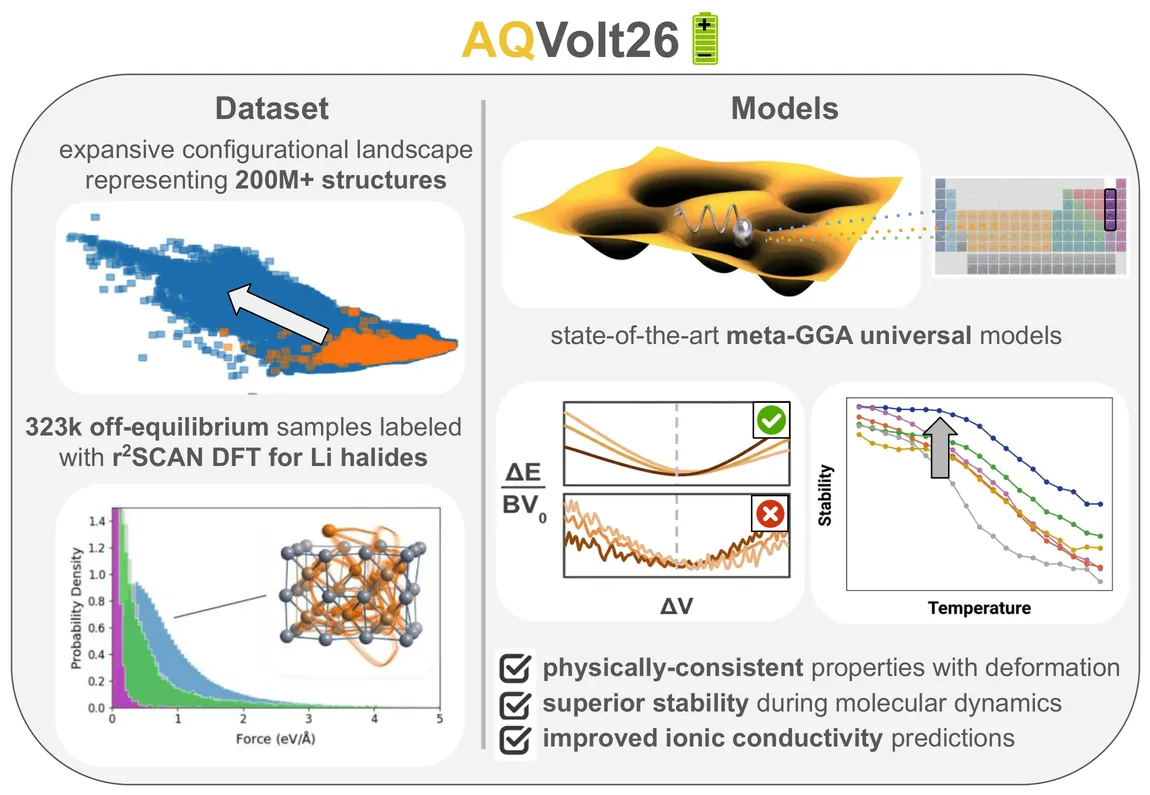
X-ray Photoelectron Spectroscopy (XPS) is a powerful technique to probe chemical states and interfacial processes in battery materials, but a quantitative interpretation is often hindered by the complex, heterogeneous microstructures that form during operation and dominate electrochemical cycling. Silicon based anodes represent a paradigmatic example in Li batteries, as (de)lithiation proceeds through the formation of strongly disordered Li$_x$Si phases and crystal-amorphous transformations that are hard to characterize. Here, we introduce a computational framework that combines machine-learning (ML) prediction of core-level binding energies to large-scale atomistic simulations -- Grand Canonical Monte Carlo (GCMC) complemented with molecular dynamics (MD), driven by a ML potential -- for a systematic sampling of lithiation states and local atomic environments. This approach yields stoichiometry maps that match the characteristic experimental trends observed in operando and ex situ XPS measurements, including the distinctive Si $2p$ spectroscopic signatures associated with the crystal-to-amorphous disordering driving early delithiation.
2602.23028
Feb 2026Materials Science
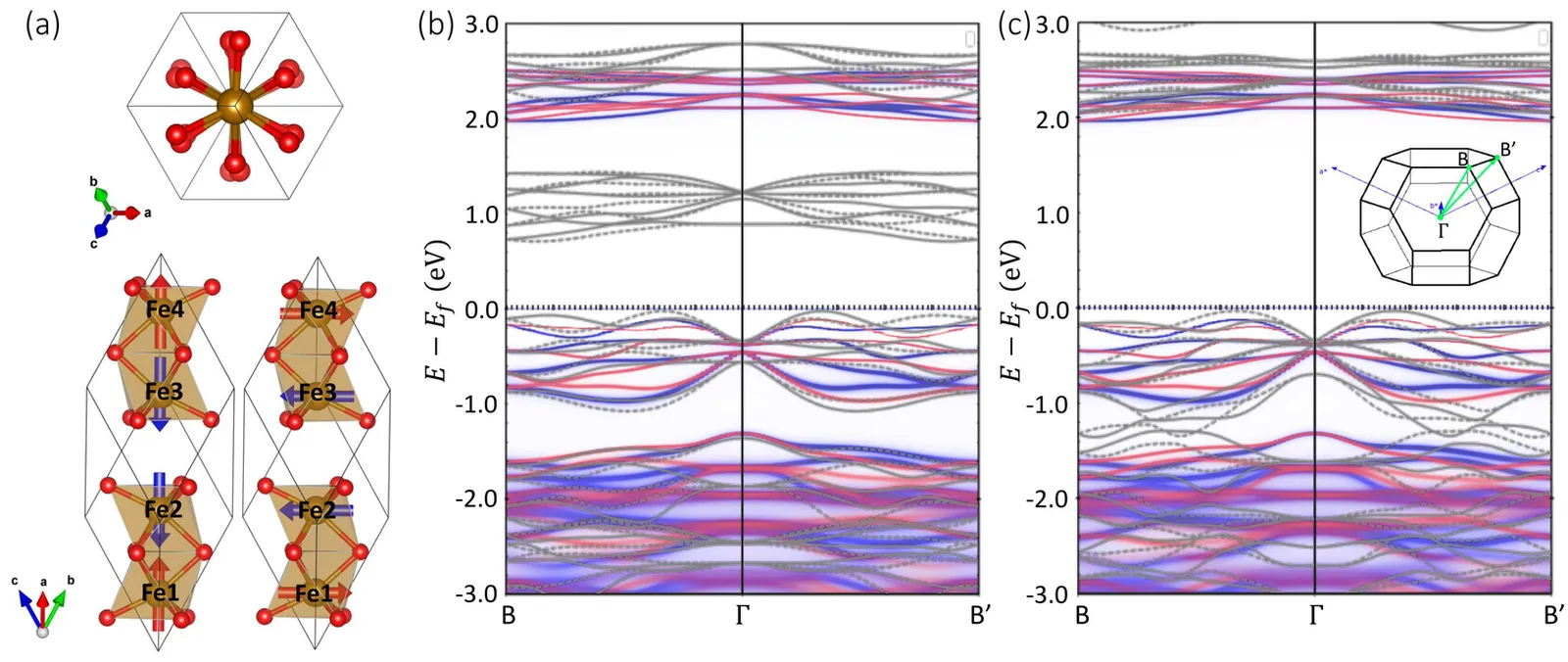
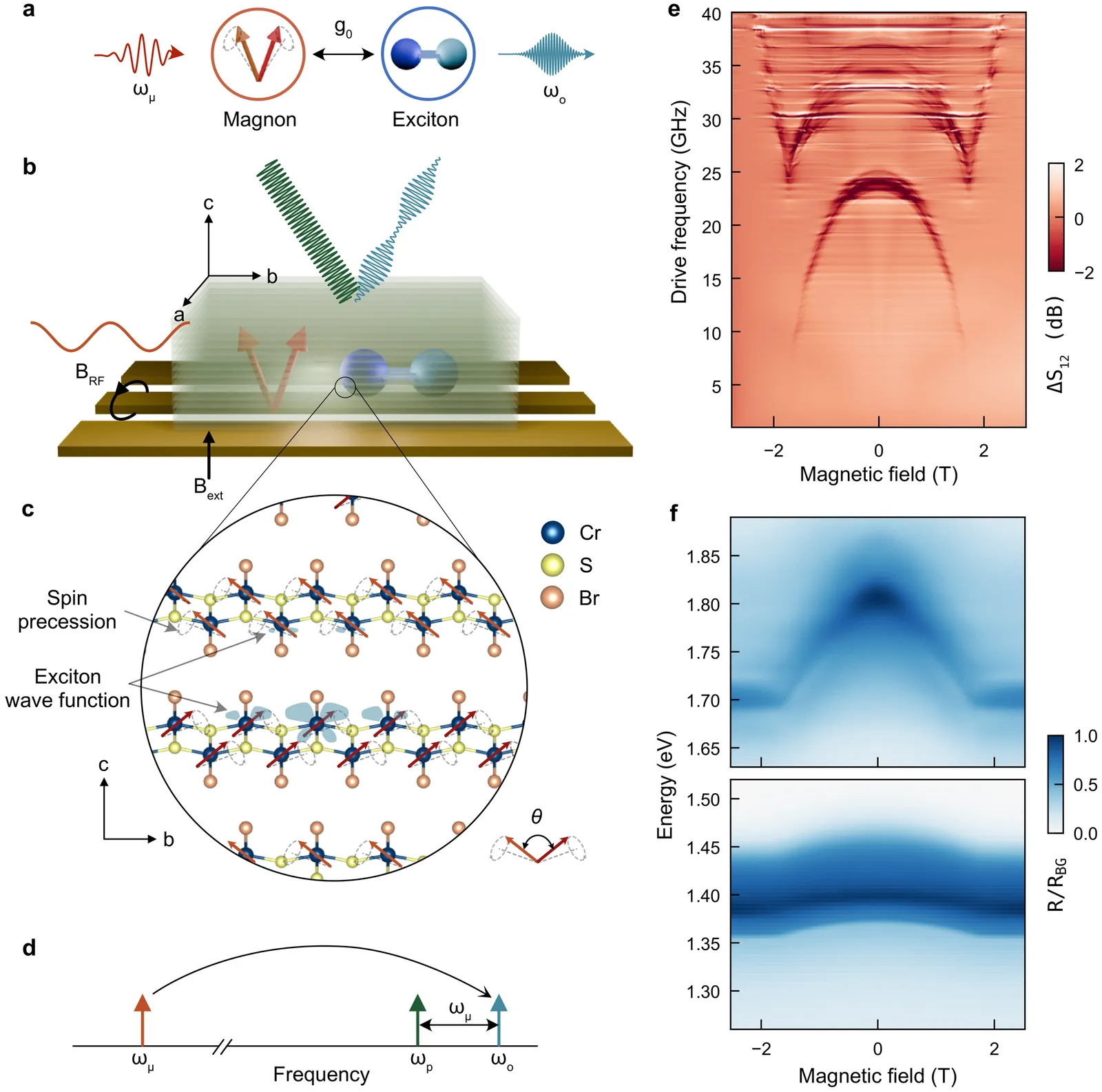
X-ray magnetic circular dichroism of altermagnet $α$-Fe$_2$O$_3$ based on multiplet ligand-field theory using Wannier orbitals
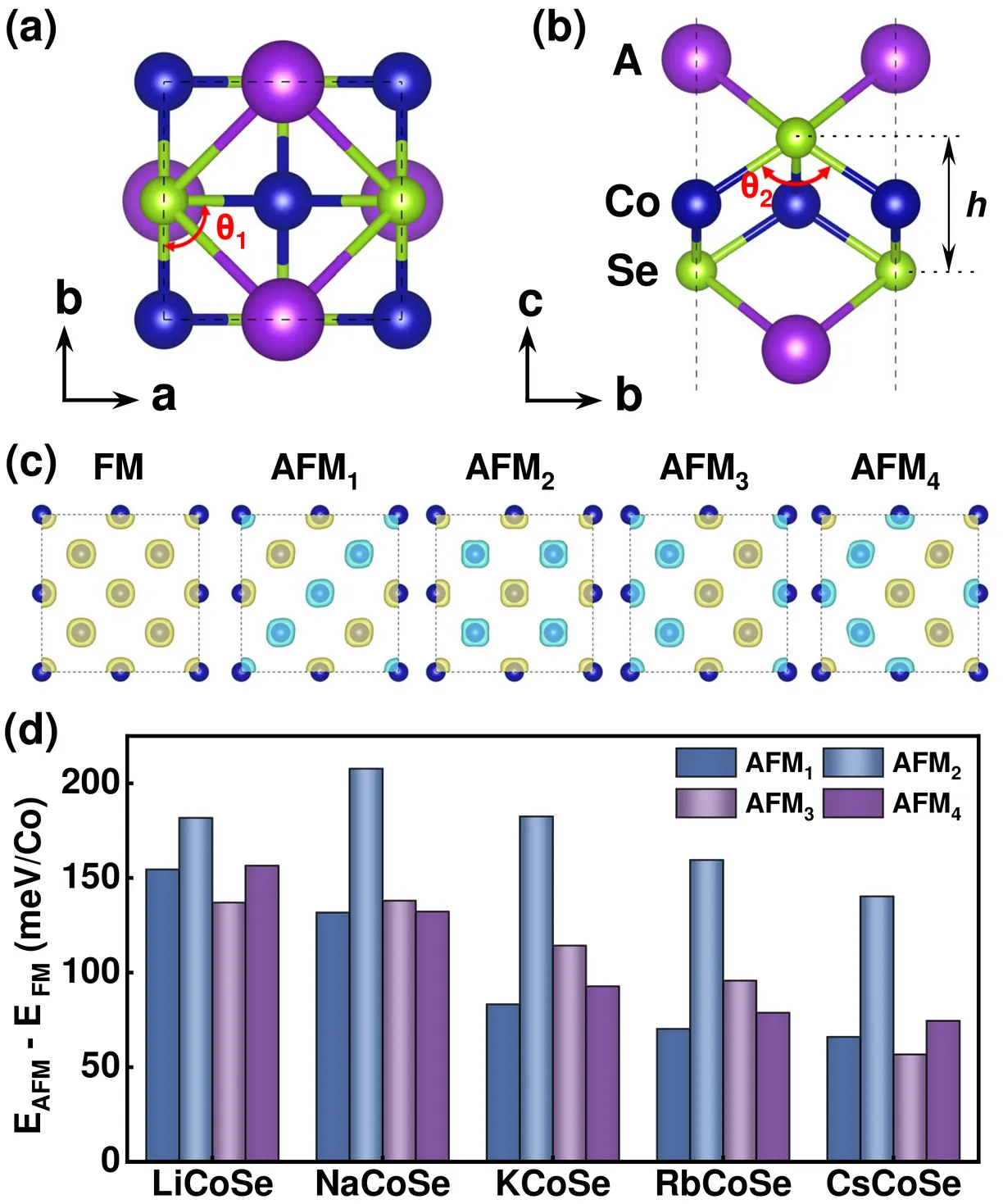
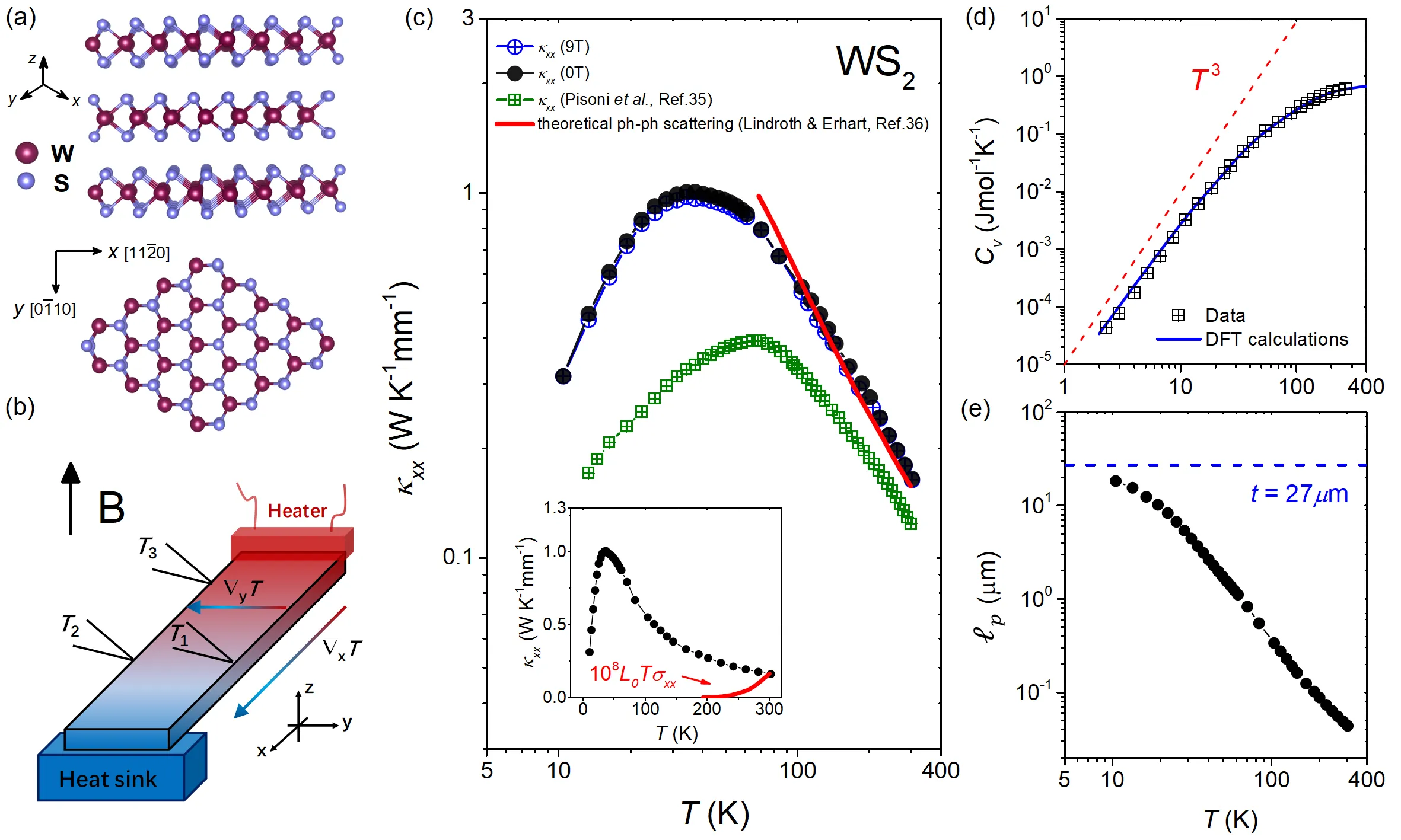
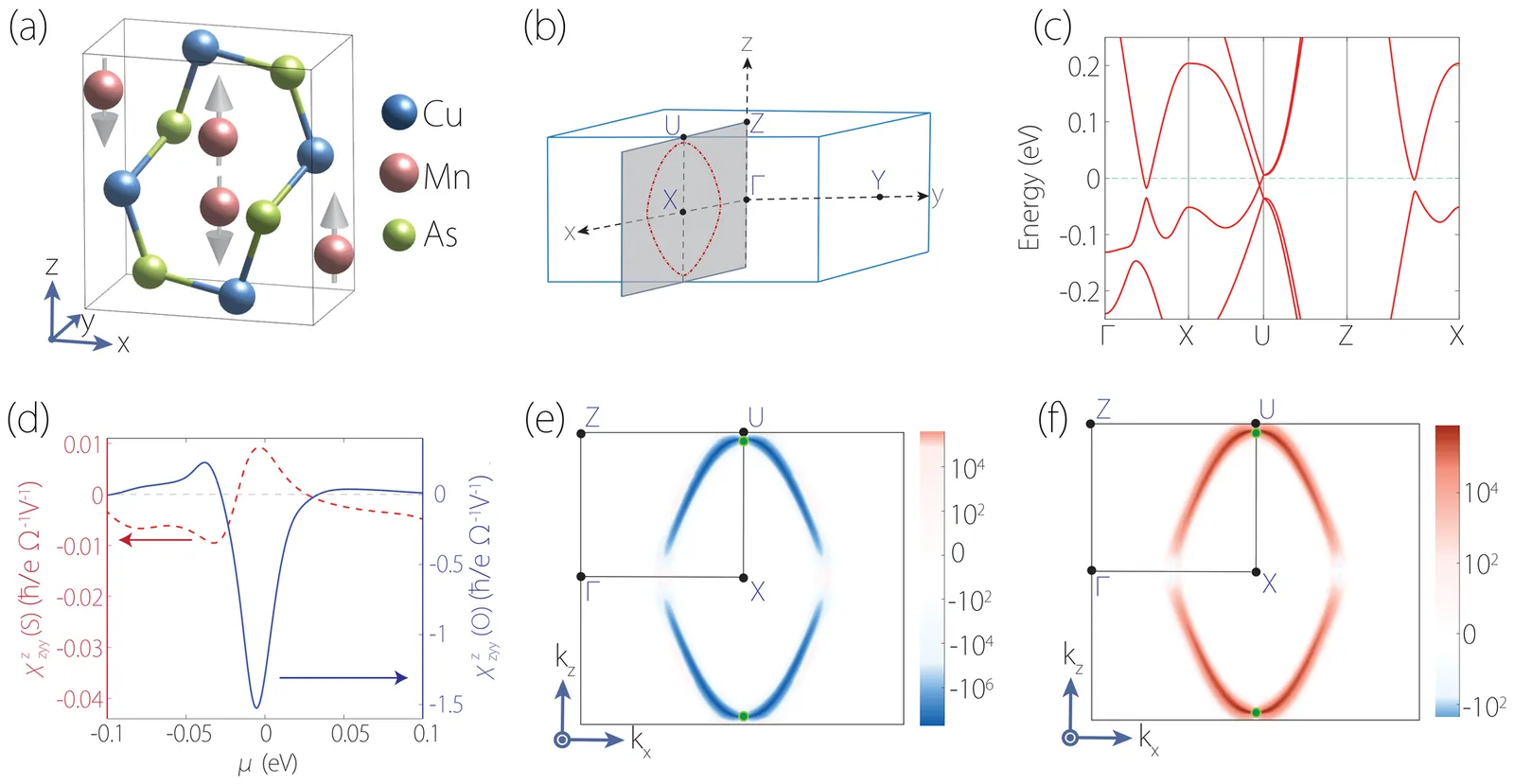
Hematite $α$-Fe$_2$O$_3$ is a $g$-wave altermagnetic material, which has an easy-axis phase and easy-plane weak ferromagnetic phase below and above Morin transition temperature, respectively. The presence of these phases renders it a good candidate to study the characteristic spin splitting in altermagnets under the impacts of relativistic effect and finite temperature. In this regard, we have calculated the band structure of $α$-Fe$_2$O$_3$ based on density functional theory (DFT) which also considers the Hubbard-U correction and spin-orbit coupling (SOC) effects. Additionally, the charge self-consistent DFT + dynamical mean-field theory (DMFT) calculations have been performed at finite temperatures. It is found that the altermagnetic spin splitting in $α$-Fe$_2$O$_3$ preserves with either SOC or temperature effect taken into account. Furthermore, we present a numerical simulation of the x-ray magnetic circular dichroism (XMCD) of the L$_{2,3}$ edge of Fe using a combination of DFT with multiplet ligand-field theory (MLFT). In terms of the different Néel vectors present in $α$-Fe$_2$O$_3$, we calculate the x-ray absorption spectroscopy (XAS) of the L$_{2,3}$ edge of Fe in the form of conductivity tensor and analyze the XMCD response from a perspective of symmetry. A characteristic XMCD line shape is expected when the Néel vector is along [010] direction (magnetic point group $2^\prime/m^\prime$) and the light propagation vector is perpendicular to the Néel vector, which can be further distinguished from the XMCD response originated from weak ferromagnetism with the light propagation vector parallel to the Néel vector.
2512.11664
Dec 2025Materials Science
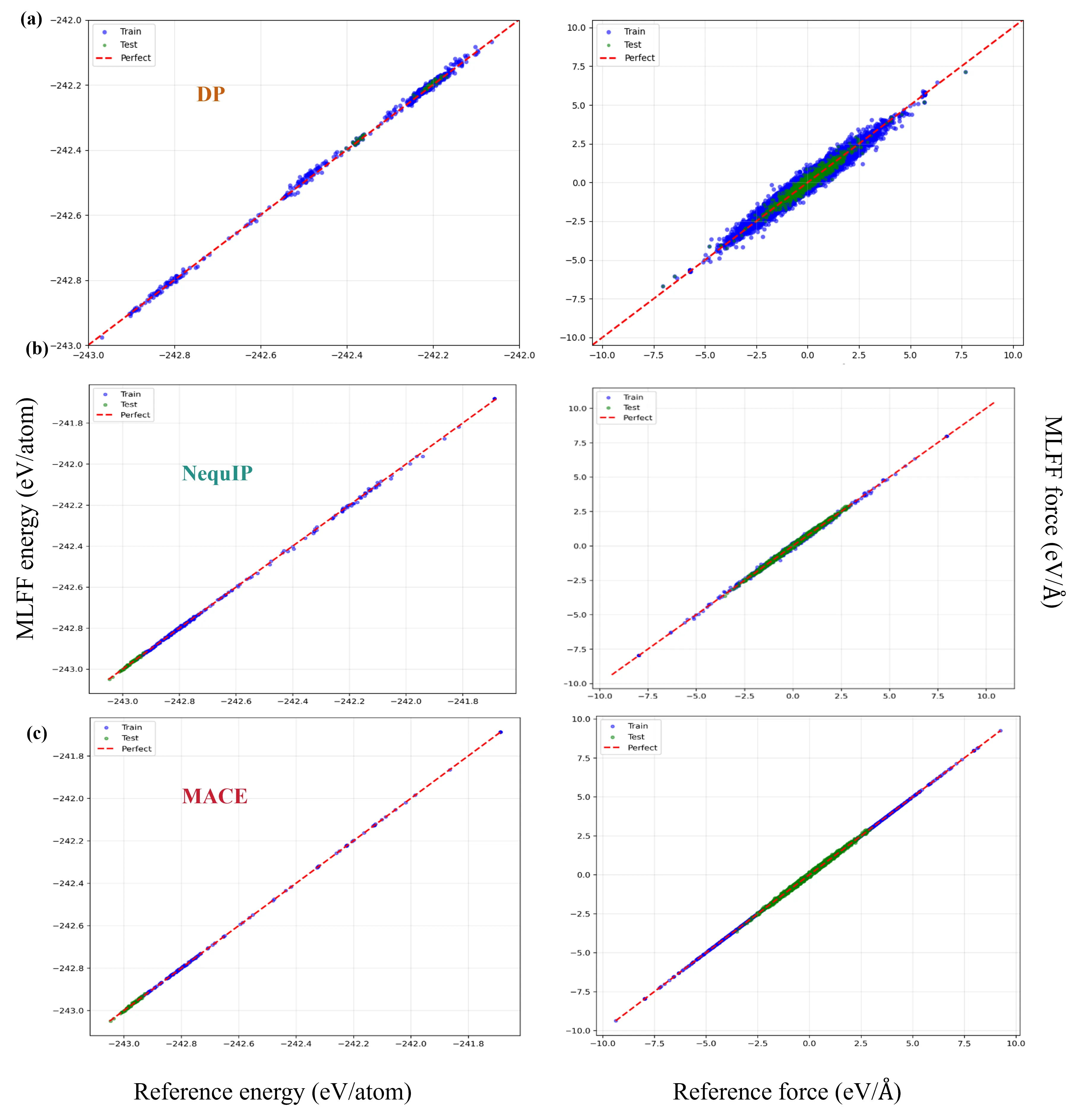
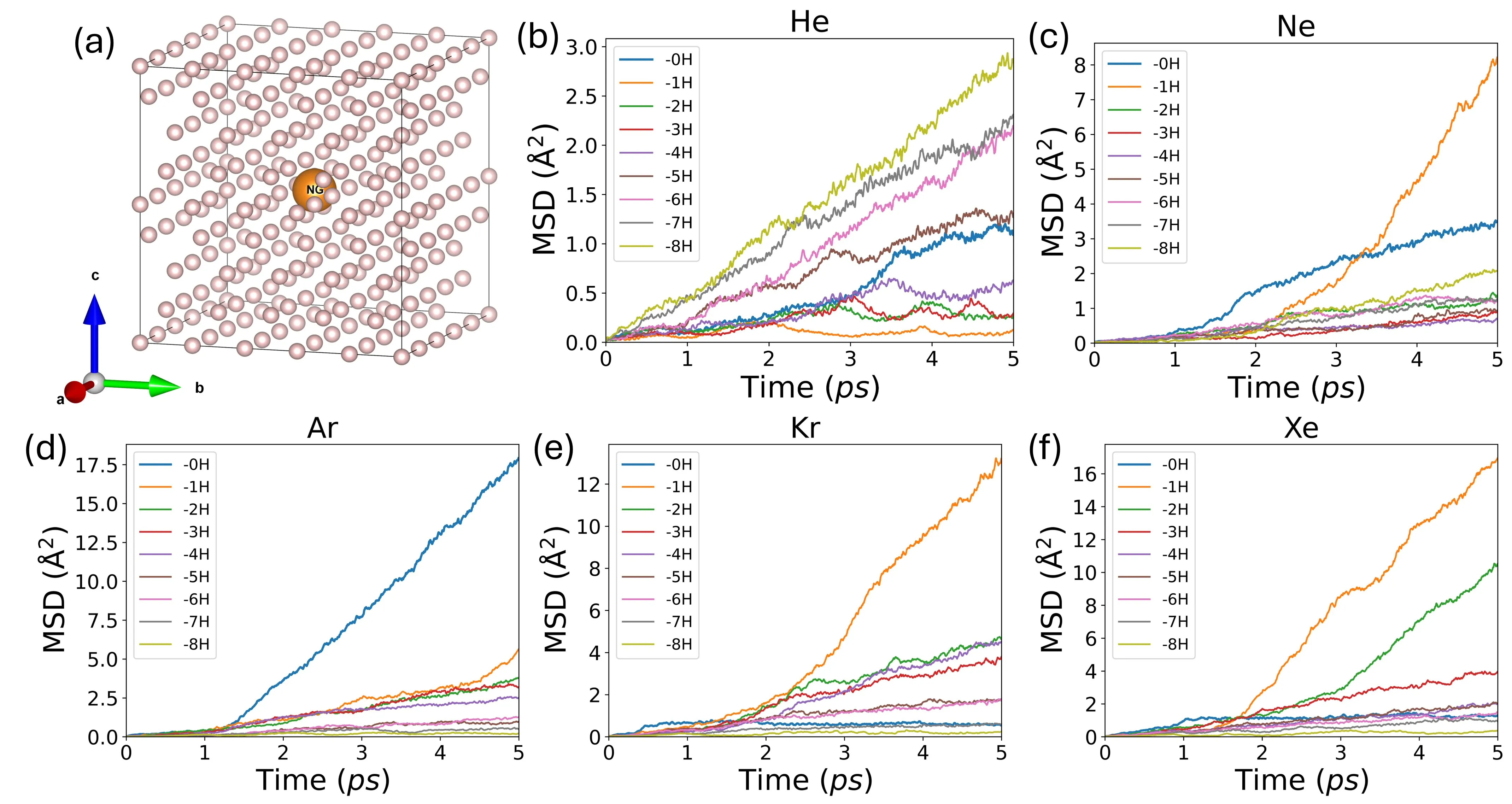
Influence of Exchange-Correlation Functionals and Neural Network Architectures on Li$^+$-Ion Conductivity in Solid-State Electrolyte from Molecular Dynamics Simulations with Machine-Learning Force Fields
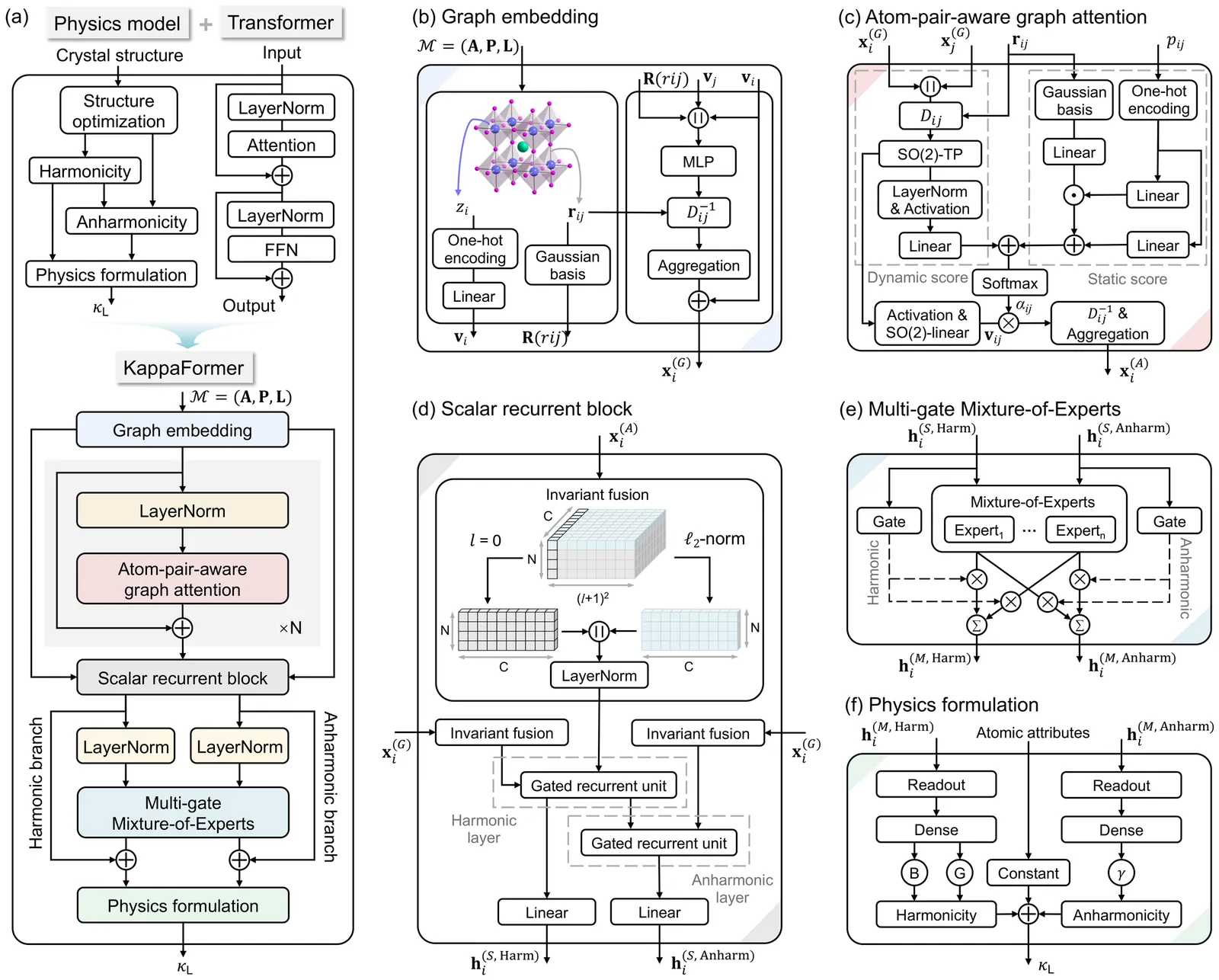
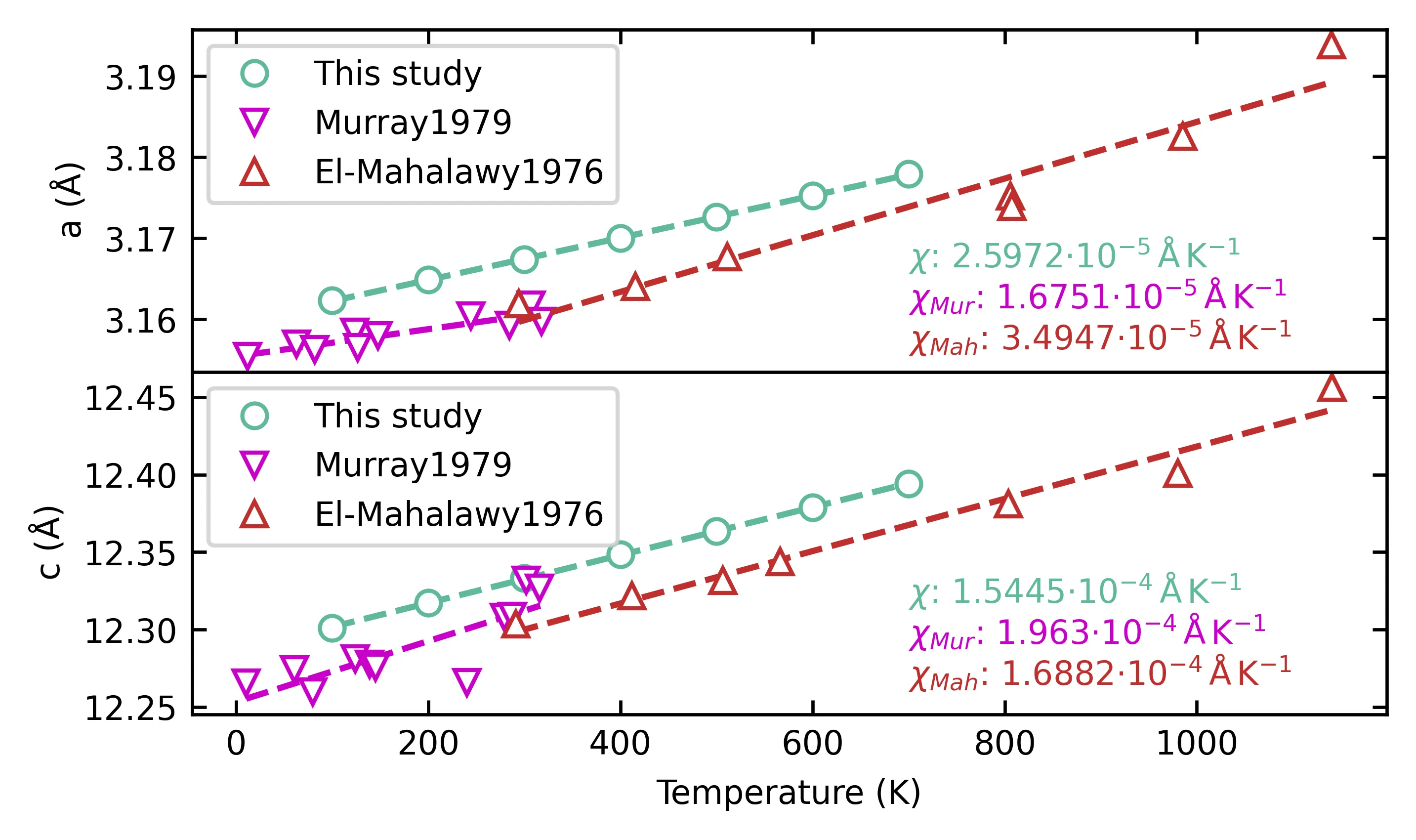
With the rapid advancement of machine learning techniques for materials simulations, machine-learned force fields (MLFFs) have become a powerful tool that complements first-principles calculations by enabling high-accuracy molecular dynamics simulations over extended timescales. Typically, MLFFs are trained on data generated from density functional theory (DFT) using a specific exchange-correlation (XC) functional, with the goal of reproducing DFT-level properties. However, the uncertainties in MLFF-based simulations--arising from variations in both MLFF model architectures and the choice of XC functionals--remain insufficiently understood. In this work, we construct MLFF models of different architectures trained on DFT data from both semilocal and hybrid functionals to describe Li$^+$ diffusion in the solid-state electrolyte Li$_6$PS$_5$Cl. We systematically investigate how different XC functionals influence the Li$^+$ diffusion coefficient. To reduce statistical uncertainty, the mean squared displacements are averaged over 300 independent molecular dynamics (MD) trajectories of 70 ps each, yielding statistical variations below $1\%$. This enables a clear assessment of the respective influences of the functional and the MLFF model. Due to its tendency to underestimate band gaps and migration barriers, the semilocal functional predicts consistently higher Li$^+$ diffusion coefficients, compared to the hybrid functional. Furthermore, comparisons among various neural network methods reveal that the differences in predicted diffusion coefficients arising from different network architectures are of the same order of magnitude as those caused by different functionals, indicating that the choice of the network model itself substantially influences the MLFF predictions. This observation calls from an urgent need for standardized protocols to minimize model-dependent biases in MLFF-based MD.
2512.11650
Dec 2025Materials Science