1,060 papers
Electron dynamics mediate the water-carbon π bond
The intermolecular interaction between a water molecule and the electrons in aromatic π systems--the water-π bond--lies at the heart of many chemical processes, yet its properties remain challenging to measure experimentally and model computationally. Infrared spectroscopy of pyrene anions hydrated by a single water molecule reveals vibrational and electronic motions that are often hidden in condensed phase measurements. Results from new machine-learning approaches to potentials and dipole moments show that the electron dynamics of the aromatic π cloud quench signals from some of water's vibrations and amplify others. The observed interplay between electronic and vibrational motions has general implications for modeling intermolecular interactions between water and aromatic systems in clusters, solutions, and at interfaces.
2604.03464Apr 2026
View
Universal Scaling and Many-Body Resurrection of Polaritonic Double-Quantum Coherences
The ultrafast nonlinear optical response of molecular ensembles is fundamentally altered under strong light-matter coupling. To rigorously isolate the genuine many-body contributions, an exact time-domain field-subtraction protocol is developed within a fully non-perturbative Maxwell-Liouville framework explicitly incorporating the two-exciton manifold in real space and time. This approach reveals that while collective cavity delocalization drives the macroscopic nonlinear signal toward a severe harmonic cancellation (an effect termed "spectral starvation"), intrinsic many-body molecular interactions robustly resurrect genuine polaritonic double-quantum coherences (DQCs). This many-body resurrection is governed by a universal two-photon matching rule, $Δ_B + 4J = Ω_R$, linking molecular anharmonicity ($Δ_B$) to the macroscopic Rabi splitting ($Ω_R$) and excitonic coupling ($J$). Crucially, this dictates that J-aggregates ($J < 0$) uniquely isolate the resonant many-body state below the dense two-exciton scattering continuum, protecting the macroscopic coherence from spatial fragmentation. This predictive framework establishes a direct phase diagram to engineer and protect optical nonlinearities across diverse strongly coupled platforms.
2604.03423Apr 2026
View
Low-Scaling Many-Body Green's Function Calculations for Molecular Systems via Interacting-Bath Dynamical Embedding Theory
We present a molecular extension of our recently proposed Green's function embedding method, interacting-bath dynamical embedding theory (ibDET), for computing charged excitation energies at the $GW$ and EOM-CCSD levels. Starting from atom-centered impurities, we construct bath representations that capture the frequency-dependent entanglement between the impurity and its environment and can be systematically improved via the construction of cluster-specific natural orbitals. Utilizing a $GW$ or coupled-cluster Green's function solver, the self-energy of the full system is assembled from all embedding problems to obtain the interacting Green's function. We show that ibDET provides accurate spectral properties with much reduced cost for a broad range of systems, including conjugated molecules and nanoclusters. Compared with full-system results, the errors in the predicted ionization potentials and electron affinities are around 0.1 eV or smaller, while each embedding problem includes only a small fraction of the total orbital space. This work provides an efficient and scalable framework for computing spectral properties of molecular systems.
2604.03137Apr 2026
View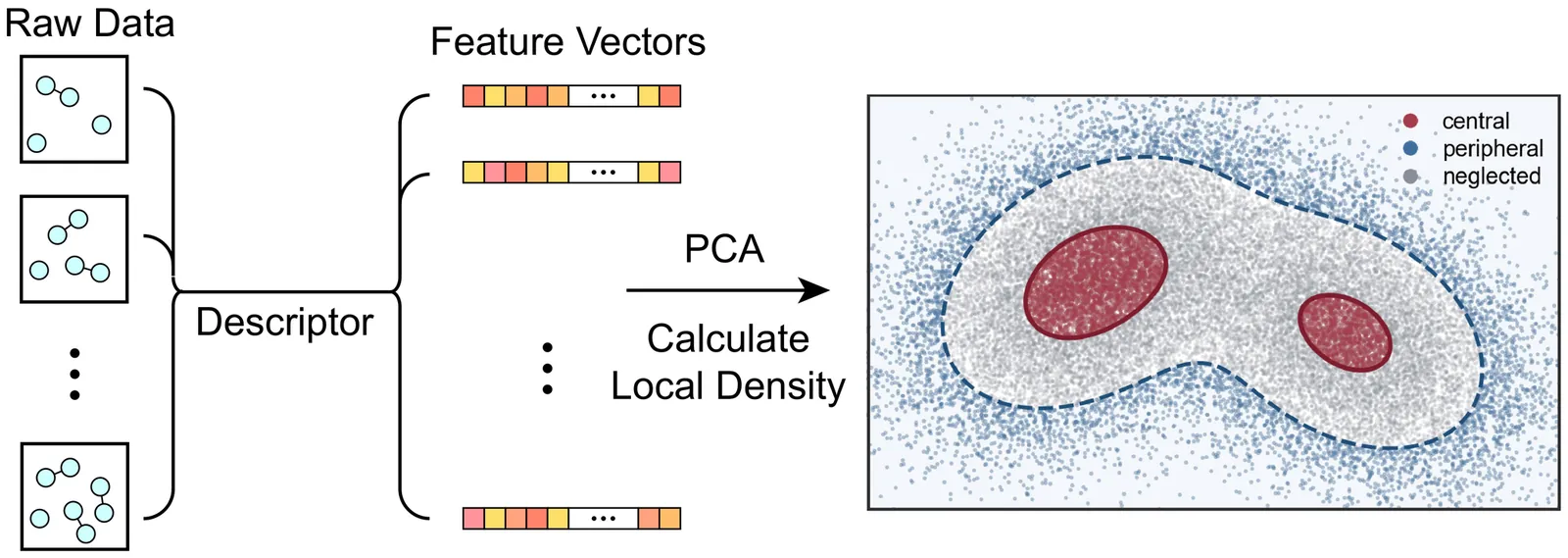
Dataset Distillation for Machine Learning Force Field in Phase Transition Regime
Machine learning force field (MLFF) has emerged as a powerful data-driven tool for atomistic simulations, enabling large-scale and complex atomic systems to be simulated with accuracy comparable to \textit{ab initio} methods. However, MLFFs often suffer from low training efficiency in the phase transition regime, where structural fluctuations are significantly elevated. To address this challenge, we propose a Central-Peripheral Distillation (CPD) algorithm for training dataset distillation. By strategically integrating representative samples with critical corner cases, the CPD algorithm ensures that the distilled dataset retains maximum structural diversity. We validated the efficacy of the CPD method on the liquid-liquid phase transition of dense hydrogen. Results show that, with the CPD approach, only 200 configurations are sufficient to train a MLFF that can fully reproduce the structural and dynamical properties of liquid hydrogen in the vicinity of its phase transition regime. This work paves the way for high-fidelity labeling of the MLFF training datasets, for instance by adopting high-level \textit{ab initio} calculations beyond the standard density functional theory, thereby enhancing the predictive accuracy of MLFFs.
2604.03027Apr 2026
View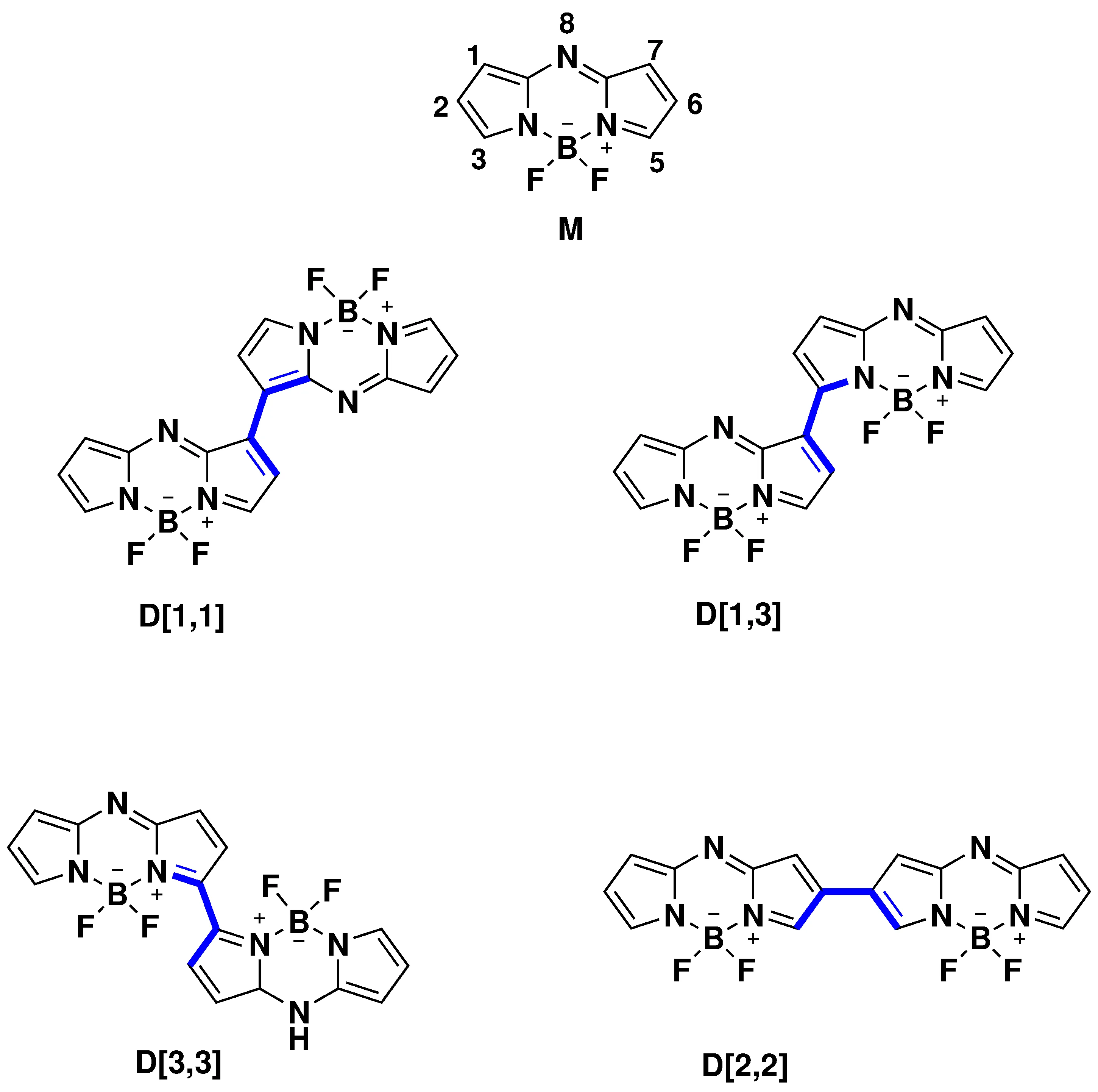
Regio-Connectivity and Torsional Angle Effects on Singlet Fission and SOCT-ISC in Aza-BODIPY Dimers
Aza-BODIPY dimers represent promising molecular systems for efficient triplet-state generation through either intramolecular-singlet fission (iSF) or spin-orbit charge transfer intersystem crossing (SOCT-ISC). In this work, we investigate the role of molecular geometry in governing these mechanisms across four regioisomeric aza-BODIPY dimers (D[1,1], D[1,3], D[3,3], and D[2,2]) using multireference quantum-chemical calculations. Ground- and excited-state properties were analyzed at the MP2 and SA-XMCQDPT levels of theory, while diabatic couplings and spin-orbit matrix elements were evaluated to estimate iSF and SOCT-ISC rate constants, respectively. Our results reveal that triplet formation is strongly governed by the torsional angle (Φ) between monomer units, with regio-connectivity exerting a secondary influence. Dimers D[1,1] and D[1,3] exhibit favorable iSF energetics and coupling magnitudes, whereas D[2,2] displays low iSF rate constant (kSF ) but enhanced SOCT-ISC activity. The D[3,3] dimer shows exothermic multiexciton formation but reduced iSF efficiency due to destructive coupling interactions. The dominant ISC channel proceeds through the S1-T3 transition with large spin-orbit coupling and a small energy gap. These findings provide critical mechanistic insights into geometry-dependent triplet generation in aza-BODIPY dimers.
2604.03011Apr 2026
View
The correlation discrete variable representation revisited
The correlation discrete variable representation (CDVR) enables efficient quantum dynamics calculation with the multi-layer multi-configurational time-dependent Hartree (MCTDH) approach on general potential energy surfaces. It employs a time-dependent quadrature to compute potential energy matrix elements, thereby eliminating the need to refit the potential to a sum of products form. The non-hierarchical CDVR conserves the inherent symmetry properties of tree-shaped wavefunction representations and drastically reduces the number of grid points compared to the original hierarchical CDVR. However, it requires projection on the space spanned by the single-hole functions (SHFs) at each node of the tree, which can introduce unphysical couplings for unconverged basis sets. In this work, the non-hierarchical CDVR is revisited and a revised approach that avoids explicit projection on the single-hole space is introduced. The computational costs of the revised approach scale favorably with the number of single-particle functions (SPFs): for a tree with three edges at each node and $n$ SPFs at each edge, a n^4 scaling is achieved. Furthermore, a revised scheme that uses artificial SPFs to systematically increase the accuracy of the CDVR quadrature is presented. Computations studying the photodissociation of NOCl, the vibrational states of methyl, and the non-adiabatic quantum dynamics of photoexcited pyrazine demonstrate the accuracy and efficiency of the revised non-hierarchical CDVR. Notably, for the 24-dimensional pyrazine system the use of the CDVR does not increase the required CPU time compared to calculations utilizing the sum of products form of the vibronic coupling model.
2604.02436Apr 2026
ViewDefinitive Assessment of the Accuracy, Variationality, and Convergence of Relativistic Coupled Cluster and Density Matrix Renormalization Group in 100-Orbital Space
Accuracy, variationality, and convergence underpin the reliability of modern electronic structure methods, yet definitive benchmarks in the relativistic regime remain elusive due to the absence of numerically exact full configuration interaction (CI) references. Recent algorithmic advances in the CI framework, enabled by the small-tensor-product (STP) decomposition approach, have dramatically extended the tractable size of the configuration space, making numerically exact CI calculations feasible in large active spaces previously beyond reach. In this work, we employ the recently developed STP-CI framework to perform large-scale numerically exact CI calculations and directly benchmark relativistic coupled cluster and density matrix renormalization group methods. Definitive benchmarking of approximate relativistic electronic structure methods is ensured through the application of the gap theorem, which provides rigorous error bounds on the CI reference and establishes a controlled standard for assessing accuracy, variationality, and convergence.
2604.02144Apr 2026
View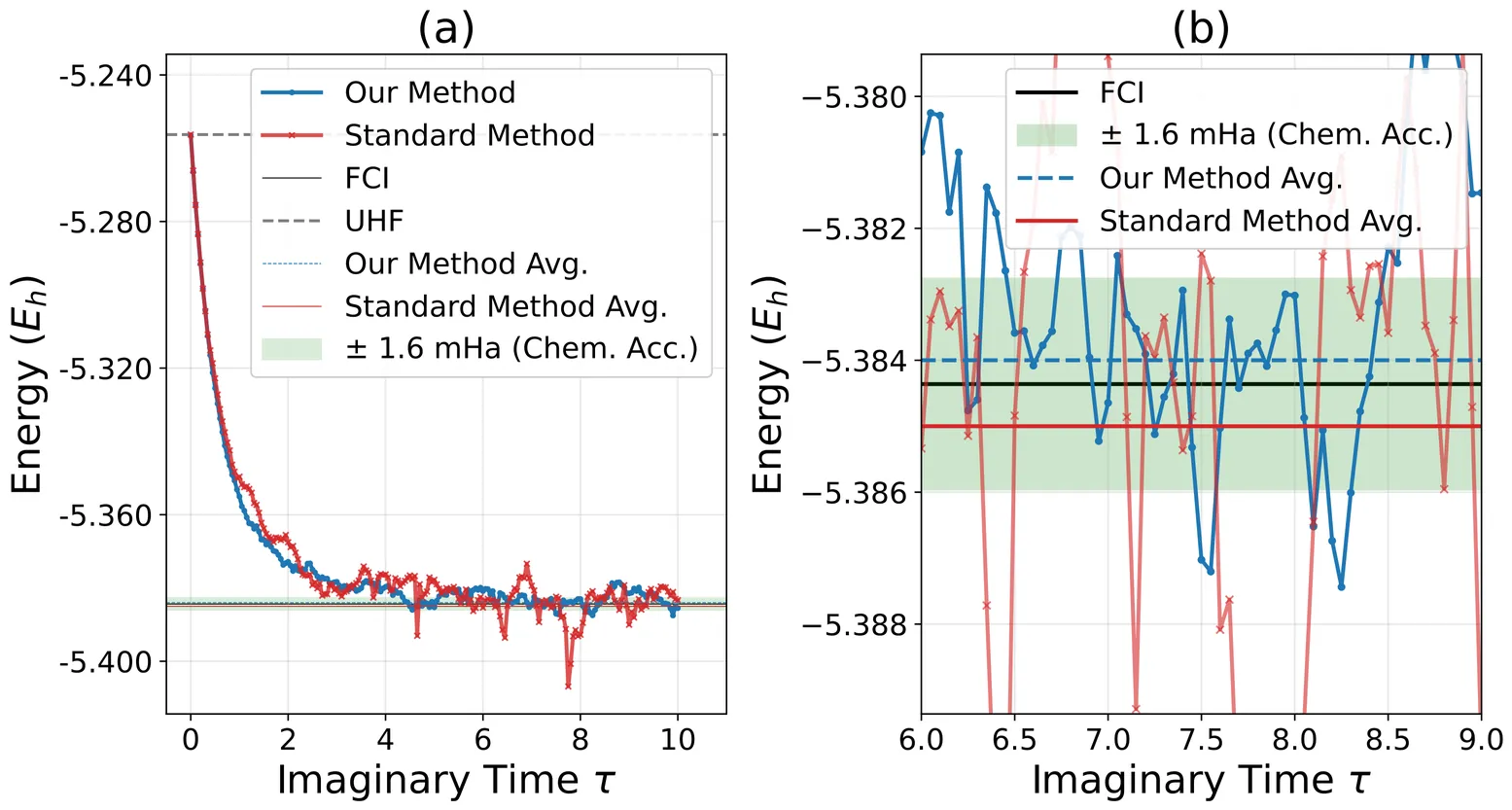
Efficient Auxiliary-Field Quantum Monte Carlo using Isometric Tensor Hypercontraction
Auxiliary Field Quantum Monte Carlo (AFQMC) has emerged as a powerful framework for treating strongly correlated electronic systems, offering a favorable balance between computational cost and accuracy. In this paper, we present a novel AFQMC method that uses the isometric tensor hypercontraction (ITHC) technique to diagonalize the two-body Coulomb interaction of molecular electronic Hamiltonians by introducing additional fictitious fermionic modes. Our method shows reduced theoretical complexity and better practical performance for both propagation and local energy evaluation compared to the standard AFQMC method. We demonstrate the efficacy of this approach by computing the ground-state energies of a linear $\ce{H10}$-chain and the benzene molecule. Our results show that the extended-basis AFQMC recovers many-body correlations with a precision comparable to that of high-level wavefunction methods such as Coupled Clusters (CC) or Density Matrix Renormalization Group (DMRG), while offering significantly improved scaling.
2604.02054Apr 2026
View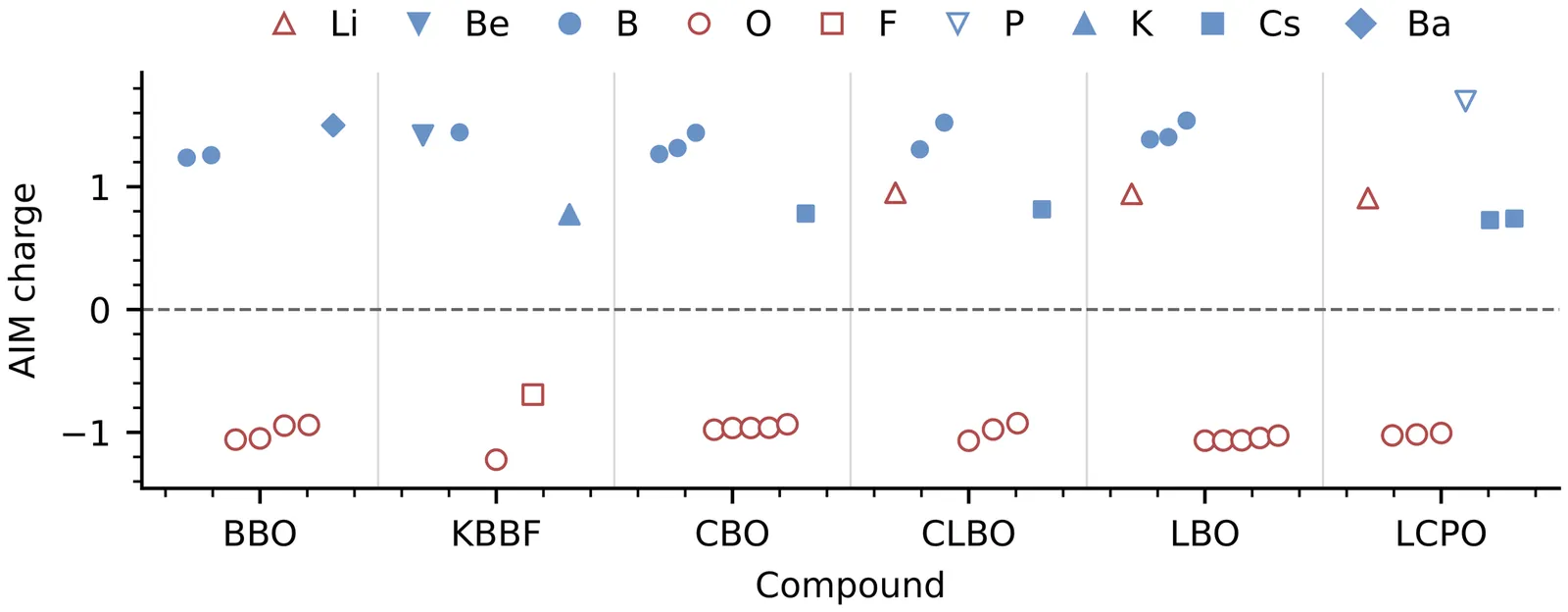
A new framework for atom-resolved decomposition of second-harmonic generation in nonlinear-optical crystals
In this work, we develop a new framework for computing atom-resolved contributions to optical properties based on atoms-in-molecules (AIM) schemes. The formalism is independent of the specific AIM method and is made rigorous by partitioning momentum matrix elements into atomic contributions while exactly satisfying the relevant sum rules. We apply it to second-harmonic generation (SHG) in six representative UV and deep-UV nonlinear-optical crystals, namely $β$-\ce{BaB2O4} (BBO), \ce{LiB3O5} (LBO), \ce{CsB3O5} (CBO), \ce{CsLiB6O10} (CLBO), \ce{KBe2BO3F2} (KBBF), and \ce{LiCs2PO4} (LCPO). The atom-triplet decomposition reveals a clear hierarchy for the largest SHG component of each crystal. In general, two-center terms provide the leading contribution, one-center terms remain comparatively small, and fully three-center terms supply an important secondary contribution. A motif-triplet decomposition further indicates behavior dominated by the anionic framework in KBBF and LBO. In BBO, CBO, and CLBO, contributions from the anionic framework and the cation sublattice act cooperatively, although the cation contribution is crystal dependent. Moreover, cooperative contributions from the phosphate framework and the Cs sublattice are also observed in LCPO, where the O-Cs contribution is particularly significant. These results may provide a new perspective for understanding the microscopic origin of SHG in nonlinear-optical materials.
2604.01920Apr 2026
View
TUNA: A streamlined quantum chemistry program for atoms and diatomics
We present TUNA, an open-source quantum chemistry program specifically designed for atoms and diatomic molecules. Within this narrow molecular domain, a broad and consistent set of electronic structure methods and calculation types is available. Energies, optimisations, vibrational frequencies, response properties, coordinate scans and ab initio molecular dynamics trajectories can be accessed through an intuitive command-line interface. A single principle underlies TUNA: once a method can be used to evaluate the energy, all properties follow from numerical differentiation. This makes the program both a transparent teaching platform and a compact environment for benchmarking methods on diatomics $\unicode{x2014}$ among the most simple yet instructive systems in quantum chemistry. Reference implementations including density functional theory, many-body perturbation theory and coupled cluster theory, supported by detailed theoretical documentation, make TUNA an accessible foundation for developing improved methods and algorithms in electronic structure.
2604.01471Apr 2026
View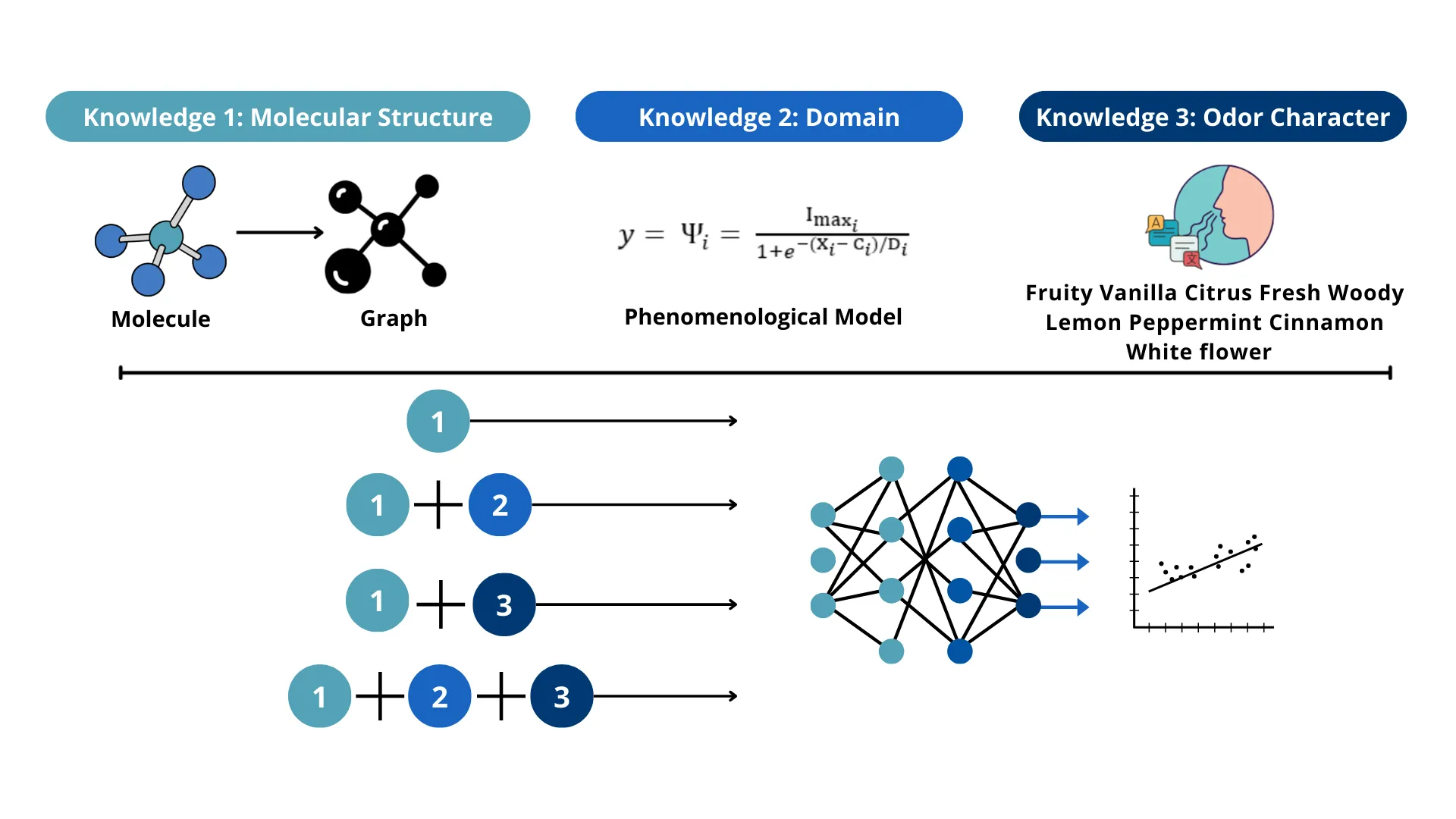
VIANA: character Value-enhanced Intensity Assessment via domain-informed Neural Architecture
Predicting the perceived intensity of odorants remains a fundamental challenge in sensory science due to the complex, non-linear behavior of their response, as well as the difficulty in correlating molecular structure with human perception. While traditional deep learning models, such as Graph Convolutional Networks (GCNs), excel at capturing molecular topology, they often fail to account for the biological and perceptual context of olfaction. This study introduces VIANA, a novel "tri-pillar" framework that integrates structural graph theory, character value embeddings, and phenomenological behavior. This methodology systematically evaluates knowledge transfer across three distinct domains: molecular structure via GCNs, semantic odor character values via Principal Odor Map (POM) embeddings, and biological dose-response logic via Hill's law. We demonstrate that knowledge transfer is not inherently positive; rather, a balance must be maintained in the volume of information provided to the model. While raw semantic data led to "information overload" in domain-informed models, applying Principal Component Analysis (PCA) to distill the 95% most impactful semantic variance yielded a superior "signal distillation" effect. Results indicate that the synthesis of these three knowledge transfer pillars significantly outperforms baseline structural models, with VIANA achieving a peak R^2 of 0.996 and a test Mean Squared Error (MSE) of 0.19. In this context, VIANA successfully captures the physical ceiling of saturation, the sensitivity of detection thresholds, and the nuance of odor character value expression, providing a domain grounded simulation of the human olfactory experience. This research provides a robust framework for digital olfaction, effectively bridging the gap between molecular informatics and sensory perception.
2604.01365Apr 2026
View
A New Paradigm for Computational Chemistry
Computational chemistry has become an indispensable tool for generating data and insights, pervading all branches of experimental chemistry. Its most central concept is the potential energy hypersurface, key to all chemistry and materials science, as it assigns an energy to a molecular structure, the necessary ingredient for reaction mechanism elucidation and reaction rate calculation. Density functional theory (DFT) has been the most important method in practice for obtaining such energies, which is mirrored in the use of high-performance computing hardware. In the last two decades, a new class of surrogate potential energy functions has been evolving with remarkable properties: quantum accuracy combined with force-field speed. Until very recently, their application was hampered by the fact that they needed to be trained on truly large system-specific data sets, generated before a computational chemistry study could be started (in sharp contrast to DFT, which, as a first-principles method, works out of the box, but at a far higher price of computational cost). Very recently, this roadblock has been overcome by so-called foundation machine learning interatomic potentials, which are poised to completely change the way we do computational chemistry, likely prompting us to abandon DFT as the prime method of choice for this purpose in less than a decade.
2604.01360Apr 2026
View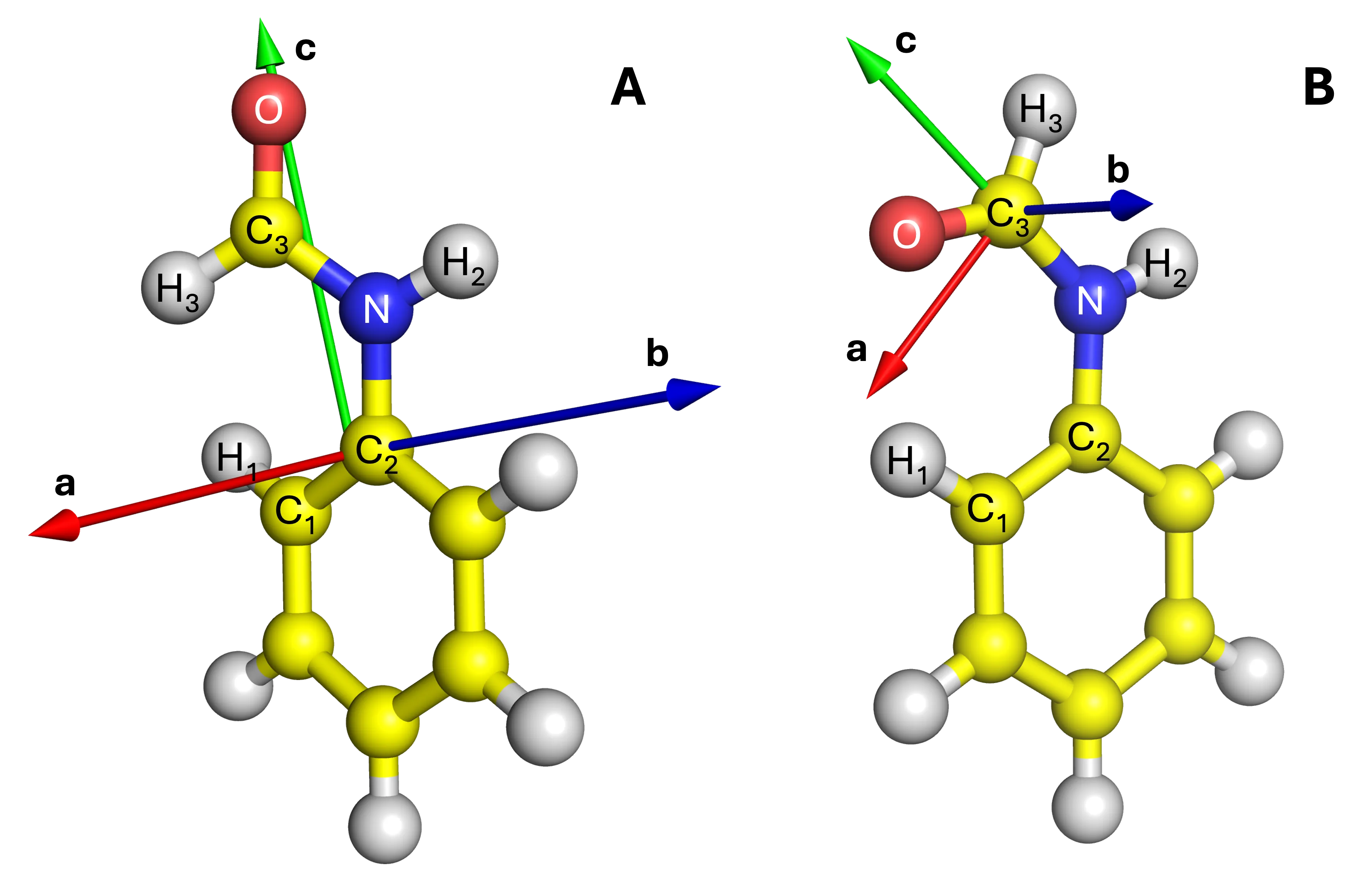
Analytic nuclear gradients including oriented external electric fields in a molecule-fixed frame
Electric field-assisted chemistry has attracted much attention in recent years, particularly in the context of oriented external electric fields for controlling molecular structure and reactivity. Such fields have been explored in a wide range of applications, including switching materials, nanoparticles, controllable catalysts, medicines, and clinical therapies. However, the use of fixed fields in the laboratory frame becomes ineffective for flexible molecules, as conformational changes can significantly alter their orientations. In this work, we propose two molecular reference frames -- the principal axis frame and the local reference frame -- to define oriented electric fields within the molecular framework. These coordinate systems powerfully eliminate ambiguities in the relative orientation between the applied field and the molecule. Analytic nuclear gradients in the presence of external electric fields are derived and implemented, with an initial application to field-dependent geometry optimizations of cis- and trans-formanilide. Analysis of the resulting field-induced equilibrium structures reveals distinct structural responses, validating the accuracy and robustness of the proposed formalism. The analytic gradient framework enables systematic investigations of molecular properties and reactivity under arbitrarily oriented electric fields, opening new opportunities for computational modeling and rational design in electric field-controlled chemistry.
2604.01189Apr 2026
View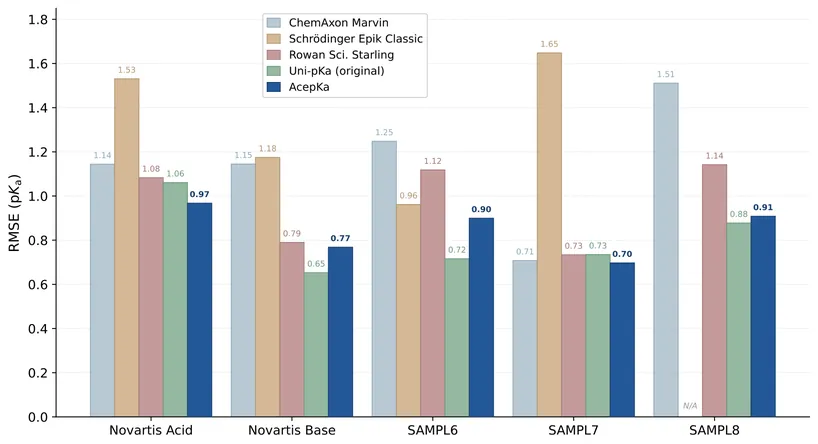
Thermodynamics-Informed Accurate pKa Prediction and Protonation State Generation in PlayMolecule AI
Accurate prediction of acid dissociation constants (p$K_{\rm a}$) and the determination of dominant protonation states is critical in drug discovery, influencing molecular properties such as solubility, permeability, and protein-ligand binding. We present Acep$K_{\rm a}$, an advanced application integrated into the PlayMolecule AI platform. Acep$K_{\rm a}$ is built upon the theoretically rigorous Uni-p$K_{\rm a}$ framework, which unifies statistical mechanics with representation learning. By modeling the complete protonation ensemble rather than treating p$K_a$ as a scalar regression target, Acep$K_{\rm a}$ ensures thermodynamic consistency across coupled ionization sites. We describe the application's enhanced architecture, which features a retrained Uni-Mol backbone achieving state-of-the-art performance on standard benchmarks. Furthermore, we detail critical engineering advancements. These include AceConfgen, a proprietary GPU-accelerated conformer generator that achieves a ~40x speed-up compared to NVIDIA's nvmolkit, a streamlined inference engine to directly protonate molecules, and a 3D-aware modality for applying protonation states to bound ligand poses. Finally, we discuss the integration of Acep$K_{\rm a}$ into the PlayMolecule AI ecosystem, a modern AI-assisted environment for molecular modelling and drug discovery.
2604.00841Apr 2026
View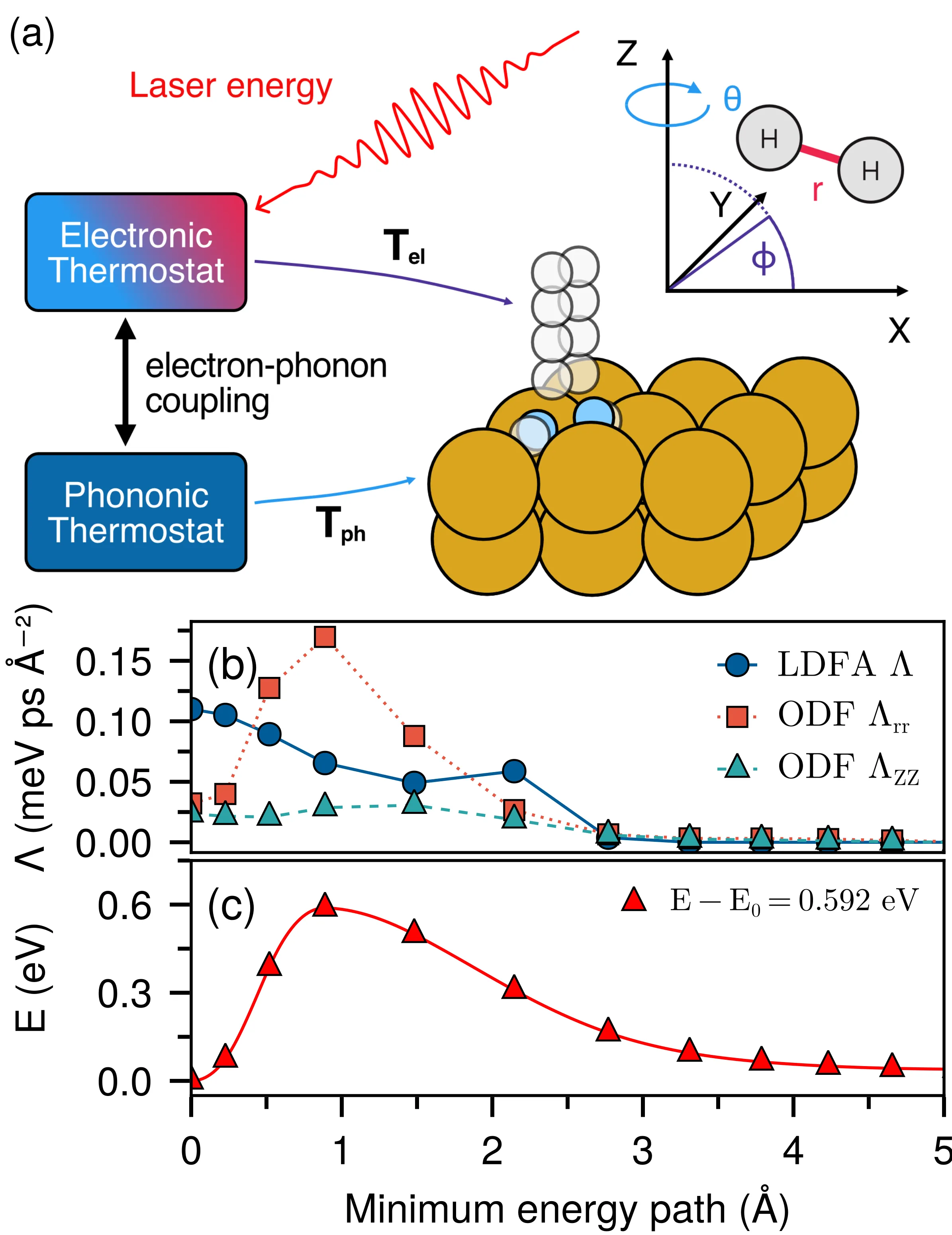
Role of anisotropic electronic friction in laser-driven hydrogen recombination on copper
Ultrafast light-driven chemical dynamics at surfaces are governed by energy transfer from excited electrons to vibrational degrees of freedom. When this nonadiabatic energy transfer is anisotropic, it can lead to dynamical steering effects that affect reaction probabilities or non-thermal final energy distributions in molecules. Here, we use a machine-learning-enabled simulation framework to compare isotropic and anisotropic models of electronic friction during laser-driven hydrogen evolution on the (111) facet of copper. While anisotropic friction strongly determines the rate of energy transfer into the adsorbate and the fluence dependence of reaction probabilities, it has little effect on final translational, vibrational and rotational energy distributions as these are mainly governed by the potential energy landscape at the barrier.
2604.00709Apr 2026
View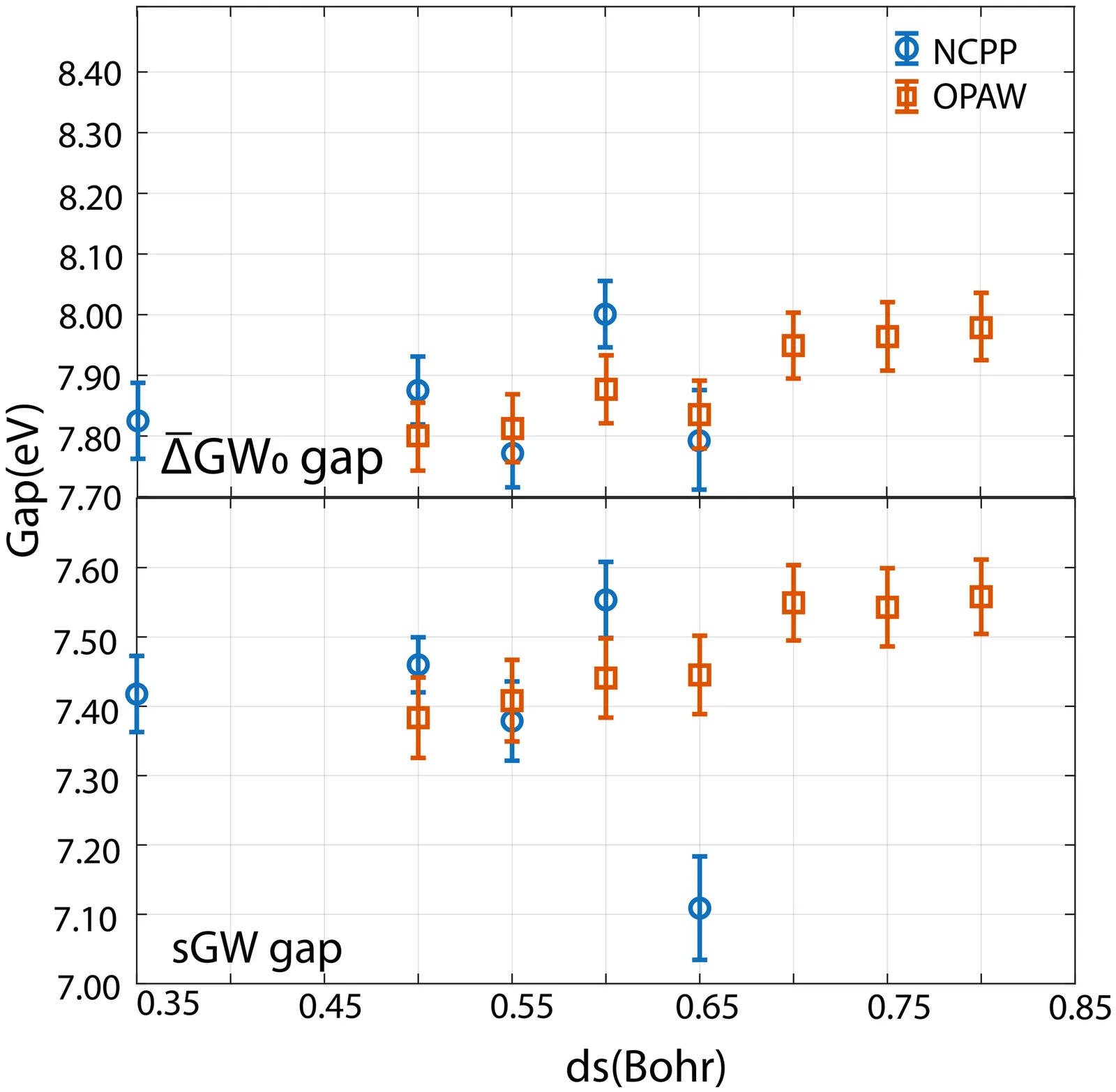
Stochastic GW with the Orthogonalized Projector Augmented Wave Method
We introduce stochastic GW with the orthogonalized projector augmented-wave method (OPAW-sGW). This implementation enables accurate quasiparticle band gaps on significantly coarser real-space grids than norm-conserving pseudopotential sGW (NCPP-sGW). The orthogonalized PAW representation preserves the formal all-electron character and enables stochastic sampling of the Green's function and screened Coulomb interaction.
2604.00522Apr 2026
View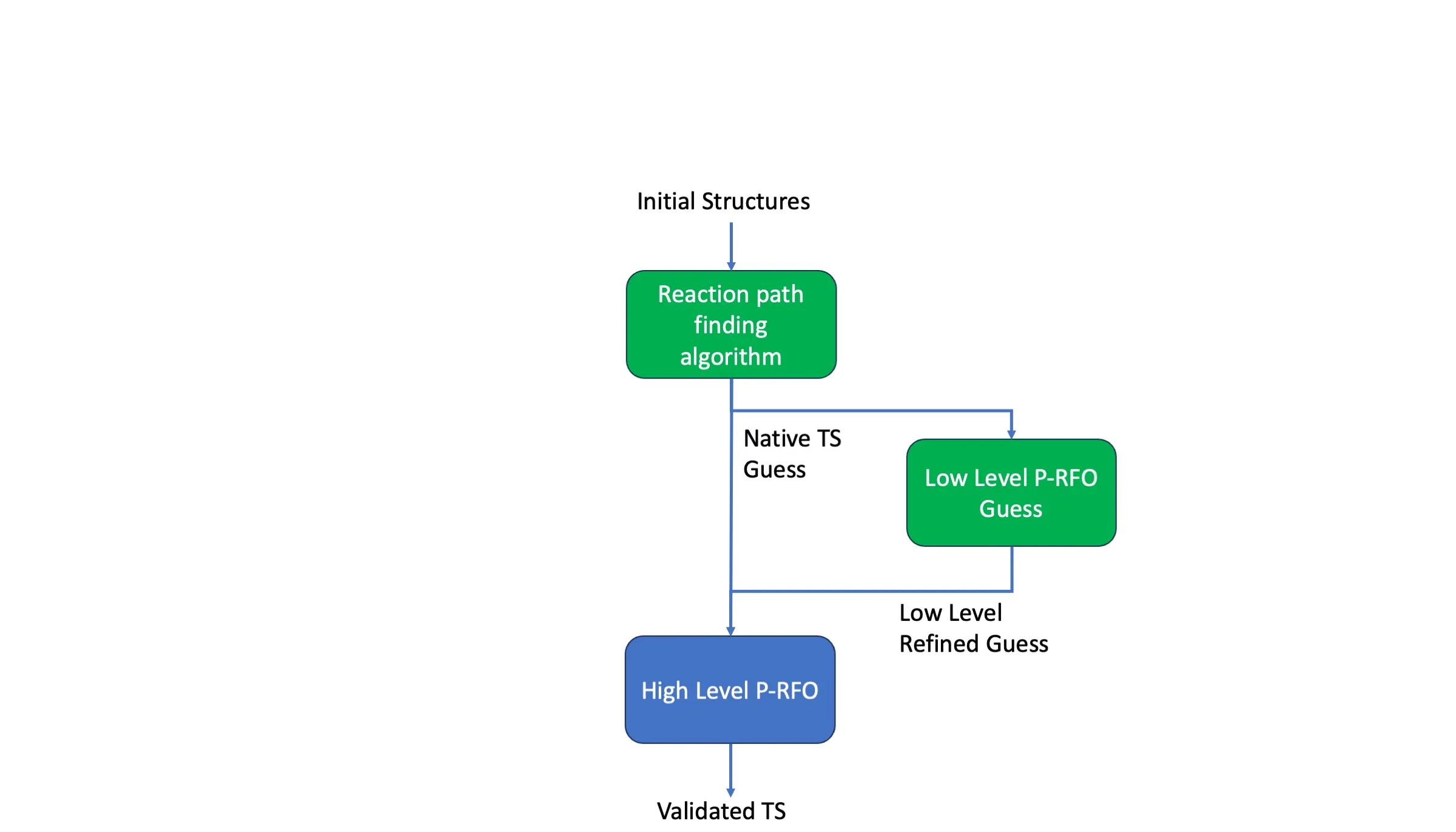
Reliable and Efficient Automated Transition-State Searches with Machine-Learned Interatomic Potentials
Transition-state searches are central to understanding reaction mechanisms, but the high computational cost of density-functional theory (DFT) limits their application in high-throughput catalyst and materials discovery. Machine-learned interatomic potentials (MLIPs) offer near-DFT accuracy at orders-of-magnitude lower cost, yet their reliability for transition-state searches remains underexplored. Here, we systematically benchmark hybrid transition-state-search workflows combining six freely available potentials (MACE-OMol25, UMA-Small, UMA-Medium, eSEN-S, AIMNet2, and GFN2-xTB) with two reaction-path-finding algorithms (the freezing-string method and climbing-image nudged elastic band) across 58 diverse reactions spanning small organics, polymerization chemistry, and transition-metal catalysis. We find that models trained on the Open Molecules 2025 dataset exhibit markedly superior performance, with MACE-OMol25 achieving a 96.6% success rate while requiring fewer than four DFT-gradient evaluations per reaction on organic systems - a 94-96% reduction compared to conventional DFT-based searches. Low-level refinement on the MLIP surface before high-level DFT optimization reduces computational cost three-fold with minimal loss in reliability. For transition-metal systems, UMA-Medium demonstrates promising transferability to in-distribution transition metal complex reactions and out-of-distribution organometallic C-H activation. These results establish MLIP-accelerated workflows as practical tools for automated reaction discovery, enabling near-DFT accuracy at a fraction of traditional expense.
2604.00405Apr 2026
View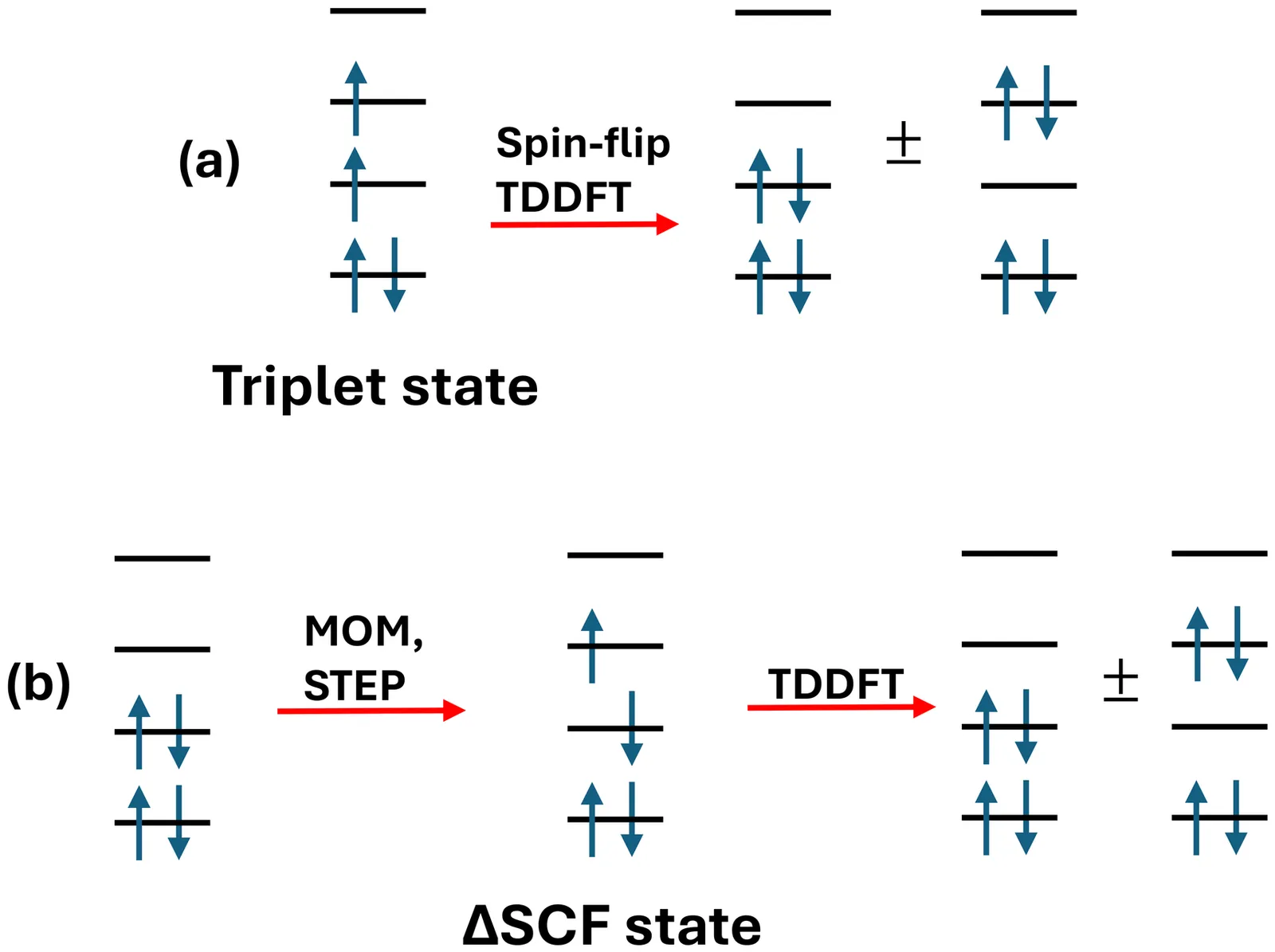
TD$Δ$SCF: Time-Dependent Density Functional Theory with a Non-Aufbau Reference for near-degenerate states
Near-degenerate electronic structures remain a major challenge for conventional single-reference density functional theory (DFT). To address this problem, we propose time-dependent $Δ$SCF (TD$Δ$SCF), a novel linear-response scheme in which a non-Aufbau $Δ$SCF determinant serves as the reference for a subsequent TDDFT calculation. In contrast to collinear spin-flip (SF)-TDDFT, this formulation preserves the usual Coulomb and exchange-correlation response contributions while describing the target states from an electronically excited reference. We examine the performance of TD$Δ$SCF for several prototypical problems involving near-degeneracy, including the torsional potential of ethylene, singlet--triplet gaps of representative diradicals, geometry optimizations of benzyne isomers, and bond-dissociation curves of hydrogen fluoride and F$_2$. Across these tests, TD$Δ$SCF shows markedly weaker functional dependence than SF-TDDFT and often yields a more balanced description of challenging singlet states. In particular, it provides smooth torsional potentials, improved singlet--triplet gaps, a consistent monocyclic structure for singlet $m$-benzyne, and a more satisfactory description of bond dissociation without the spurious low-lying states found in SF-TDDFT. At the same time, the method exhibits a systematic tendency to overestimate singlet energies and can lose accuracy when the underlying $Δ$SCF reference is not well suited to the final state. We also identify a numerical instability that can arise in non-Aufbau calculations and trace its origin to the exchange-correlation potential near uncompensated nodal regions. These results highlight both the promise and the practical limitations of TD$Δ$SCF as a low-cost method for singlet states with near-degenerate electronic structures.
2603.29948Mar 2026
View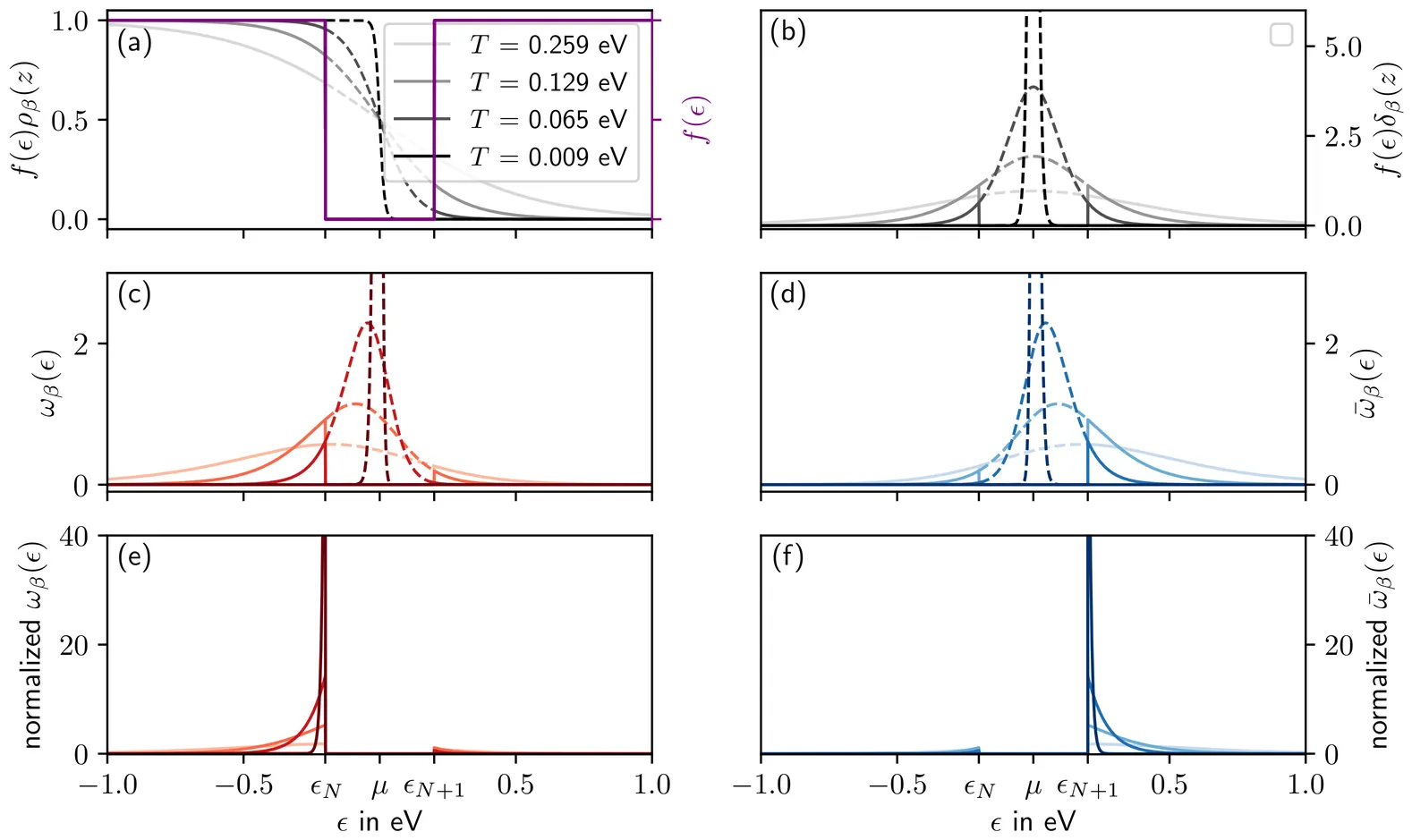
Gap edge eigenpairs from density matrix purification using moments of the Dirac distribution
In this work, we propose a simple method to resolve the eigenstates located at the band gap edges of an electronic eigenspectrum using only the quasi-purified one-particle density matrix as input. The theoretical framework relies on the decomposition of the occupation number variance into a particle and hole moment. These moments, when purified using power narrowing iterations, allow to isolate the higher occupied and lower unoccupied single state projectors, giving readily access to the corresponding eigenpairs. We demonstrate that when degeneracy is encountered, power narrowing remains able to deliver relevant mixed states. From a benchmark of selected molecules, we show that the method is robust and efficient since it requires no more that a dozen of matrix-matrix multiplications at worst. The possibility of reducing the computational cost using Lanczos subspace approach is discussed. The very low algorithmic complexity of power narrowing makes it very easy to implement in electronic structure codes or libraries already featuring Fermi operator expansion or density matrix purifications.
2603.29849Mar 2026
View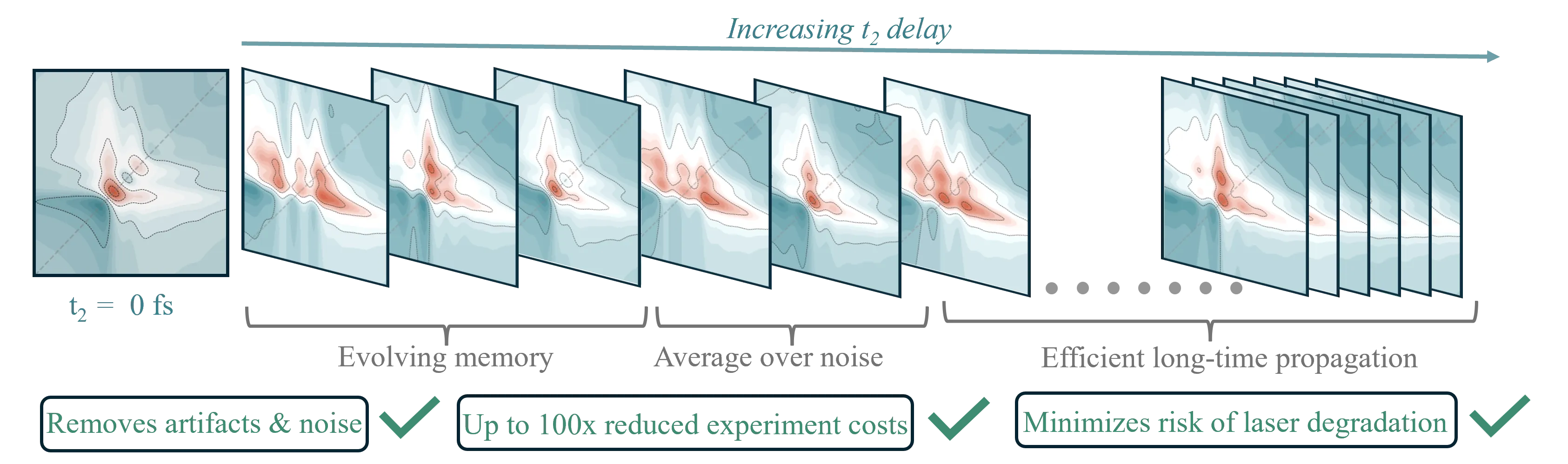
Short-lived memory in multidimensional spectra encodes full signal evolution
Ultrafast multidimensional spectroscopies are powerful tools that can access charge and energy flow in complex materials, shifting chemical kinetics, and even many-body interactions in correlated matter. However, current implementations typically involve complex apparatuses and long averaging times. As a result, these methods have been limited to detailed mechanistic investigations of a few samples, precluding the broad characterization of molecular systems and/or the optimization of material ones. For example, converging the statistical noise in 2D spectra becomes exponentially expensive with increasing waiting times, and extended laser exposure heightens the probability of sample degradation. We address this fundamental challenge by developing a new technique, the spectral generalized master equation (GME), that enables one to employ short-waiting time 2D spectra to determine the full evolution of 2D spectra over arbitrary waiting times with high temporal resolution. In addition to reducing the cost of experiments by multiple orders of magnitude, our approach accurately removes statistical noise, reducing the need for time averaging, while circumventing the increasing convergence costs with longer waiting times. We provide a rigorous theoretical footing for the spectral GME and demonstrate its applicability on theoretically generated and experimentally measured 2D electronic and 2D infrared spectra. We anticipate that this advance has the potential to enable the investigation of systems that are too delicate for study with current multidimensional spectroscopies and accelerate the progress of 2D spectroscopy-based microscopies that can offer highly resolved excitation dynamics with spatial resolution over heterogeneous environments.
2603.29814Mar 2026
ViewPage 1 of 53