15 papers

Scaling Dependencies in Irradiation-Driven Molecular Dynamics Simulations: Case Study of W(CO)$_6$ Fragmentation
Irradiation-driven fragmentation and chemical transformations of organometallic molecules play a central role in nanofabrication techniques based on the use of focused charged-particle beams. In this paper, the electron irradiation-induced fragmentation dynamics of W(CO)$_6$, a commonly used precursor for focused electron beam-induced deposition (FEBID), is investigated using the irradiation-driven molecular dynamics (IDMD) method. Simulations are performed for gas-phase systems with different precursor densities and under different irradiation conditions. The results reveal progressive fragmentation of W(CO)$_6$ molecules into W(CO)$_n$ species, accompanied by the formation of W-rich molecular clusters. The evolution of fragment abundances shows a strong dependence on both precursor density and electron fluence. Higher densities and larger fluences result in more extensive fragmentation and promote the aggregation of tungsten atoms into small metal clusters. Under certain irradiation conditions, the studied molecular systems evolve towards a steady state characterised by slightly varying fragment abundances. The obtained scaling relations between irradiation parameters and fragment distributions provide guidance for selecting simulation parameters in IDMD simulations of the FEBID process, ensuring a quantitative description of precursor fragmentation dynamics.
2603.25064Mar 2026
View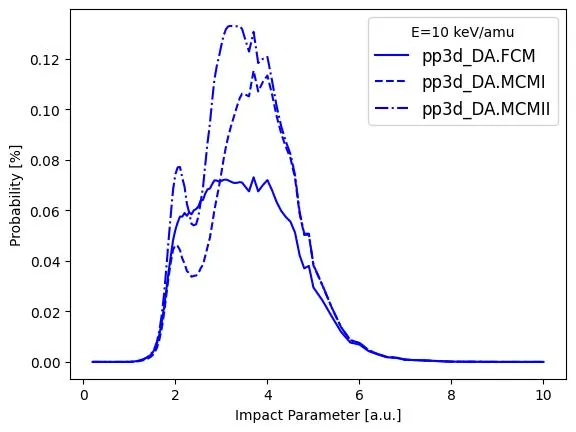
Interatomic Coulombic decay initiated by electron removal and excitation processes in helium ion and argon dimer collisions
The electron removal and excitation channels in argon dimer target and helium ion projectile collision systems that facilitate interatomic Coulombic decay (ICD) are investigated. We implement an independent-atom and independent-electron model of the collision system with the dimer target fixed at its equilibrium bond length and the He$^{2+}$ and He$^+$ ion projectiles travelling parallel to the dimer axis at impact energies ranging from 10 keV/amu to 150 keV/amu. The coupled-channel two-center basis generator method for orbital propagation is used within both a frozen atomic target approximation and a dynamic response framework. Given that ICD is facilitated through electron excitation pathways in argon dimers, a statistical technique called determinantal analysis is employed to investigate these channels. The analysis is further subdivided into models that exclude and include projectile charge changes during the collision. Electron configurations of the form Ar$^{+}$($3p^{-2}nl$) offer a pathway to ICD and are investigated, along with other one- and two-electron removal channels that lead to Ar$^{+}$-Ar$^{+}$ fragmentation. We find that the $3d$ excited state is an overall dominant channel for ICD with other excited states ranging from $4s$-$4f$ also being significant contributors. Our study notes differences between static and dynamical potential models across the projectile impact energy range, though of decreasing significance as the impact energy approaches 150 keV/amu. We also find that a He$^{+}$ projectile offers a strong pathway for ICD as the projectile impact energy decreases.
2603.22769Mar 2026
View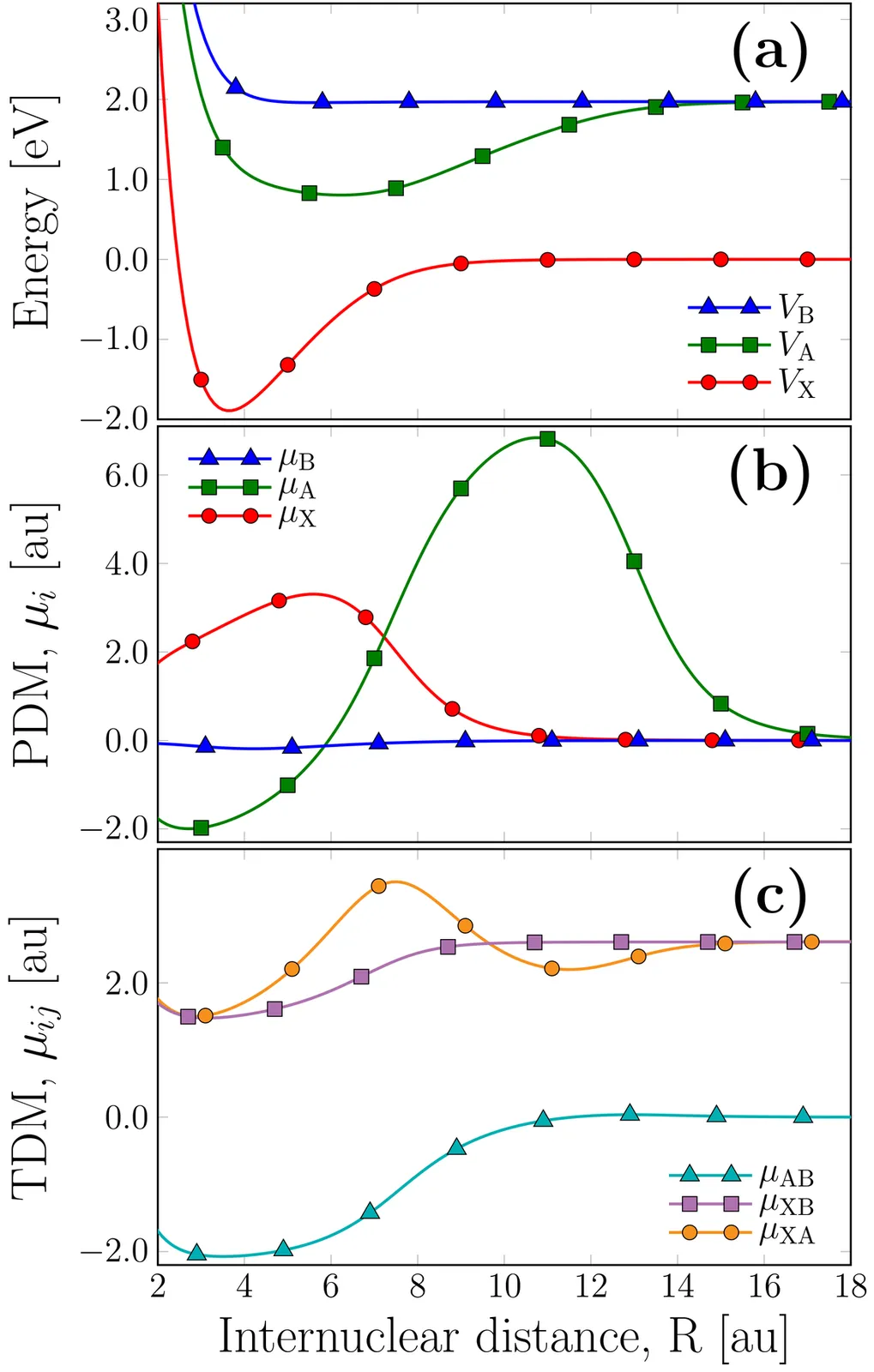
Light-induced nonadiabatic photodissociation of the NaH molecule including electron-rotation coupling
It is well established that electronic conical intersections (CIs) in molecular systems can be induced by laser light, even in diatomic molecules. The emergence of these light-induced degeneracies leads to strong coupling among electronic, vibrational, and photonic modes, which significantly influences ultrafast nuclear dynamics. In this work, we perform pump-probe numerical simulations on the NaH molecule, considering the first three singlet electronic states- (X1Σ+(X), A1Σ+(A) and B1Π(B)) -and including several light- induced degeneracies in the theoretical model. To elucidate the ultrafast molecular dynamics, the combined effects of multiple light-induced nonadiabatic couplings and rotational motion of the nuclei, together with the situation when the electronic angular momentum projected onto the diatomic axis couples with the angular momentum of the nuclei has been studied. We then calculate key dynamical observables such as dissociation probabilities, kinetic energy release spectra, and angular distributions of the photofragments within and above the linear regime.
2603.11033Mar 2026
View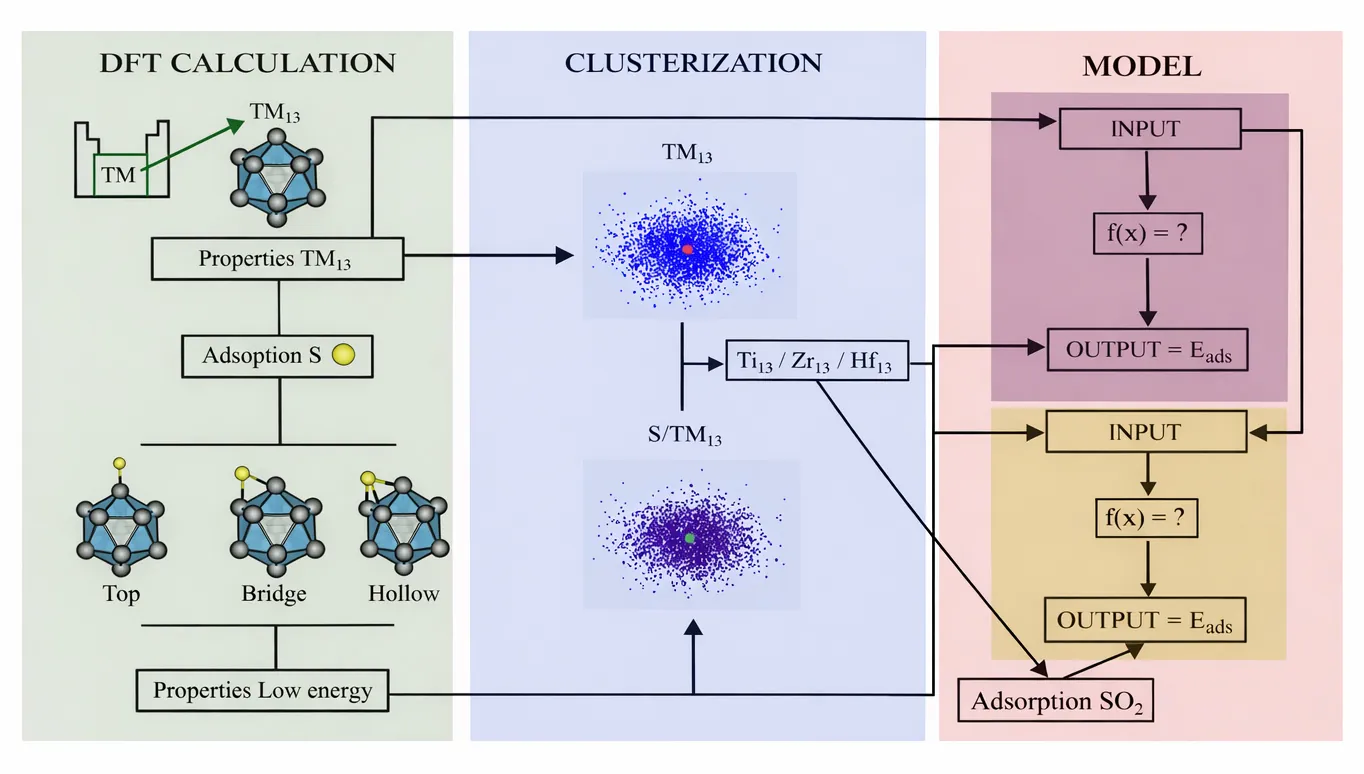
Interpretable, Physics-Informed Learning Reveals Sulfur Adsorption and Poisoning Mechanisms in 13-Atom Icosahedra Nanoclusters
Transition-metal nanoclusters exhibit structural and electronic properties that depend on their size, often making them superior to bulk materials for heterogeneous catalysis. However, their performance can be limited by sulfur poisoning. Here, we use dispersion-corrected density functional theory (DFT) and physics-informed machine learning to map how atomic sulfur adsorbs and causes poisoning on 13-atom icosahedral clusters from 30 different transition metals (3$d$ to 5$d$). We measure which sites sulfur prefers to adsorb to, the thermodynamics and energy breakdown, changes in structure, such as bond lengths and coordination, and electronic properties, such as $\varepsilon_d$, the HOMO-LUMO gap, and charge transfer. Vibrational analysis reveals true energy minima and provides ZPE-based descriptors that reflect the lattice stiffening upon sulfur adsorption. For most metals, the metal-sulfur interaction mainly determines adsorption energy. At the same time, distortion penalties are usually moderate but can be significant for a few metals, suggesting these are more likely to restructure when sulfur is adsorbed. Using unsupervised \textit{k}-means clustering, we identify periodic trends and group metals based on their adsorption responses. Supervised regression models with leave-one-feature-out analysis identify the descriptors that best predict adsorption for new samples. Our results highlight the isoelectronic triad \ce{Ti}, \ce{Zr}, and \ce{Hf} as a balanced group that combines strong sulfur binding with minimal structural change. Additional DFT calculations for \ce{SO2} adsorption reveal strong binding and a clear tendency toward dissociation on these clusters, linking electronic states, lattice response, and poisoning strength. These findings offer data-driven guidelines for designing sulfur-tolerant nanocatalysts at the subnanometer scale.
2601.13845Jan 2026
View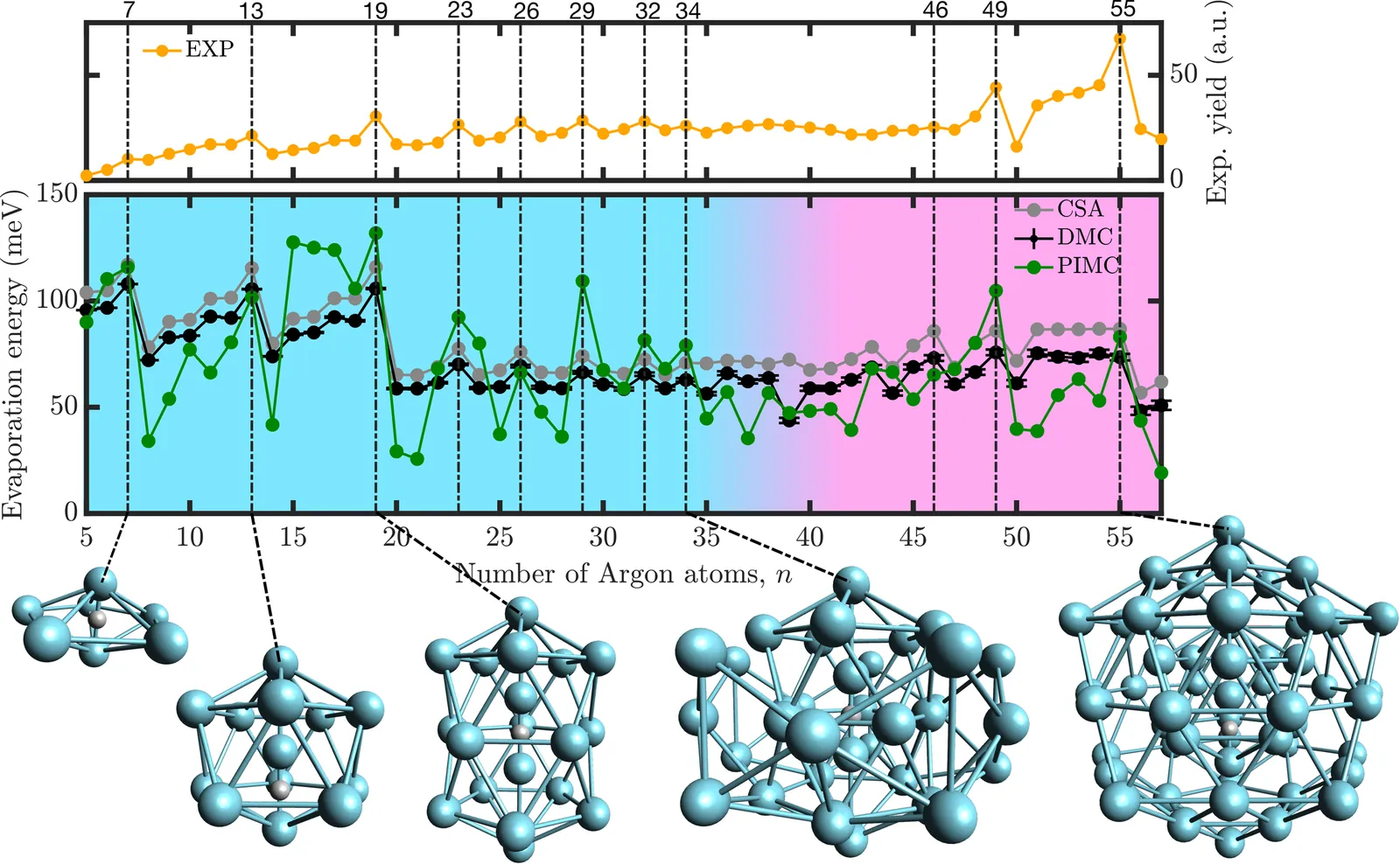
Coordination-driven magic numbers in protonated argon clusters
The structural properties of rare-gas clusters can be primarily described by a simple sphere packing model or by pairwise interactions. Remarkably, adding a single proton yields a large set of magic numbers that has remained unexplained. In this Letter, we unravel their origin by combining quantum Monte Carlo techniques with many-body ab initio potentials that correctly capture the proton's coordination environment. Thanks to this approach, we find that argon atoms are mainly localized around the classical minimum, resulting in a particularly rigid behavior in stark contrast to lighter rare-gas clusters. Moreover, as cluster size increases, we identify a clear structural transition from many-body coordination-driven stability to a regime dominated by two-body interactions, reflecting a reshaping of the underlying potential energy landscape.
2601.00591Jan 2026
View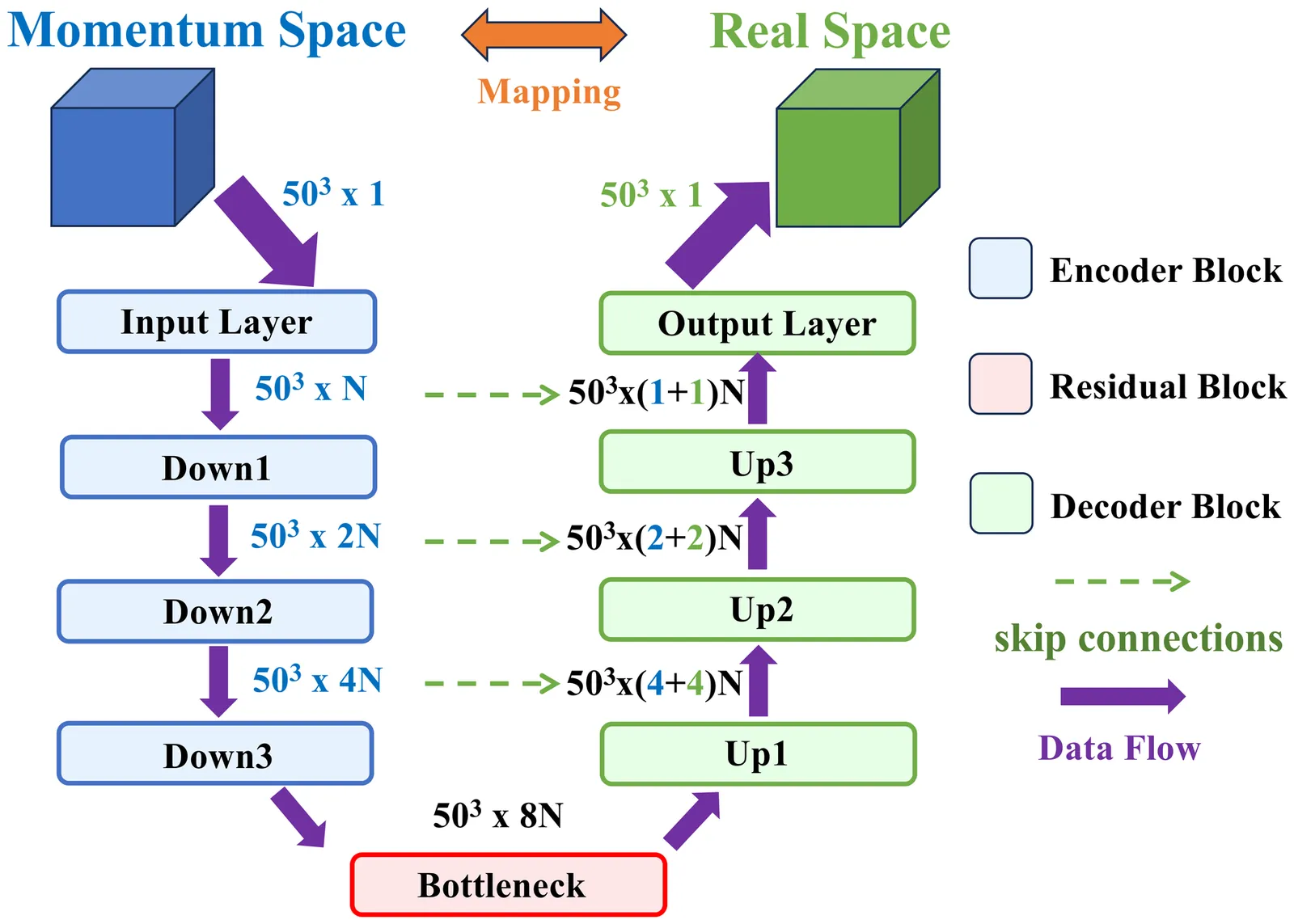
Decoding Molecular Geometries in Coulomb Explosion Imaging via Physics-Informed Deep Neural Network
Determining the absolute configuration of gas-phase molecules in position-space has long been a fundamental challenge in molecular physics. While strong-field-induced Coulomb explosion imaging (CEI) has emerged as a powerful tool for probing molecular stereochemistry in momentum-space, reconstructing the original three-dimensional structure of polyatomic molecules remains a long-standing challenge due to the inherent complexity of multidimensional inversion. Here, we introduce a deep learning framework that bridges this gap by directly recovering position-space molecular structures from Coulomb explosion momentum patterns. Our approach combines CEI simulations with a neural network trained to establish the mapping between momentum-space Newton plots and real-space geometries. The trained model demonstrates high fidelity in reconstructing the structure of CHF$_3$ from experimental CEI data. This generalizable framework can not only be extended to other molecular systems but also opens avenues for time-resolved structural analysis of molecular dynamics.
2512.16559Dec 2025
View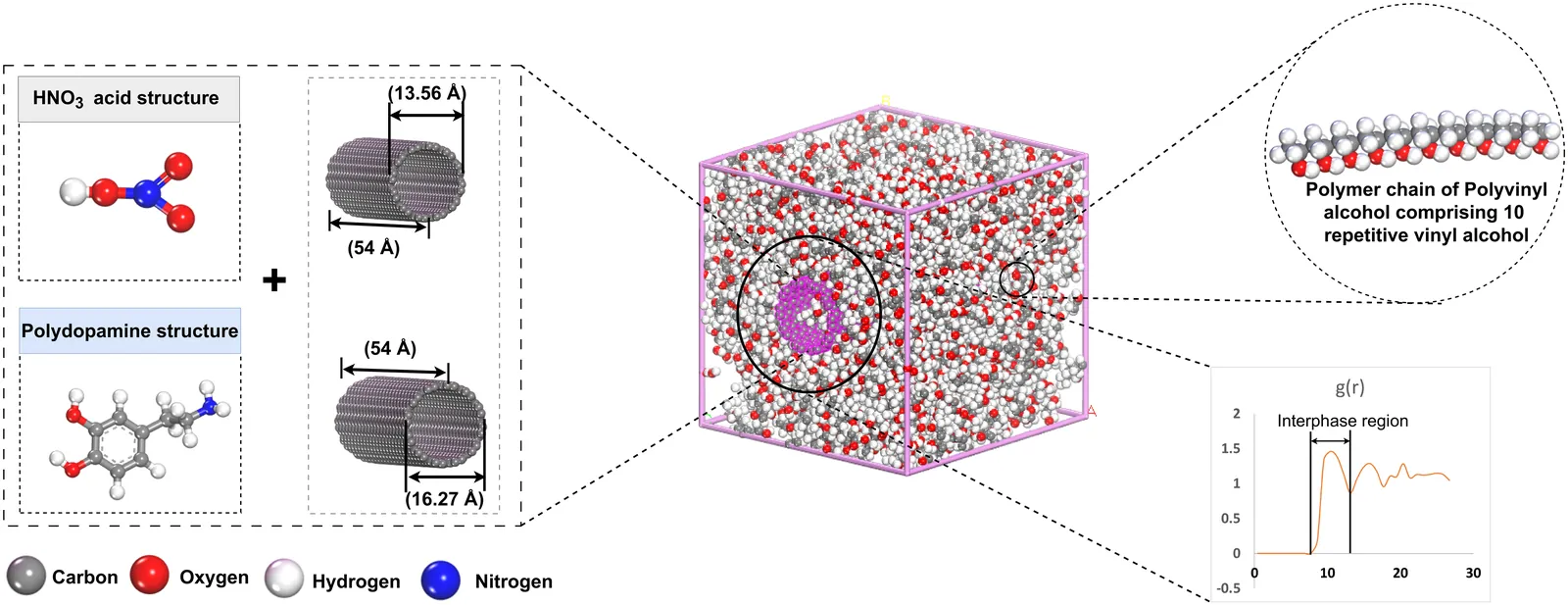
Curvature-Dependent Polarity of Interfacial Energy Flow in Functionalized CNT Polymer Nanocomposites: A Reactive Molecular Dynamics Perspective
Carbon nanotube (CNT)-polymer composites are widely engineered using surface coatings and chemical treatments to improve interfacial bonding and load transfer. It has been suggested in the nanocomposite literature that nanotube curvature, in conjunction with surface functionalization such as polydopamine (PDA) coating, could serve as an additional control knob for tuning interfacial bonding and energy dissipation in polymer-CNT systems. While experimental and simulation studies have demonstrated the benefits of PDA functionalization, the fundamental mechanism by which nanotube curvature modulates interfacial energy flow and mechanical polarity remains unresolved. This gap is sharpened by a persistent paradox: identical PDA functionalization strengthens some CNT-polymer systems while weakening others, a curvature-dependent inconsistency that has remained unexplained. Here, we employ reactive molecular dynamics (ReaxFF) simulations to resolve how curvature and PDA functionalization jointly govern interfacial energy evolution in CNT-polyvinyl alcohol (PVA) nanocomposites. Our investigation reveals that curvature and PDA functionalization jointly produce opposite regimes of interfacial energy flow: high-curvature CNTs generate dissipative, frictional interphases, whereas low-curvature CNTs confine energy in rigid, cohesive shells. This polarity inversion originates from a curvature-induced transition in PDA adsorption geometry that transforms the interphase from an energy-releasing to an energy-storing configuration. These results establish curvature as a fundamental design parameter for engineering polymer-nanotube interfaces, offering a predictive route to tune interfacial energy flow, mechanical resilience, and transport properties beyond the limits of conventional chemical functionalization.
2511.18560Nov 2025
View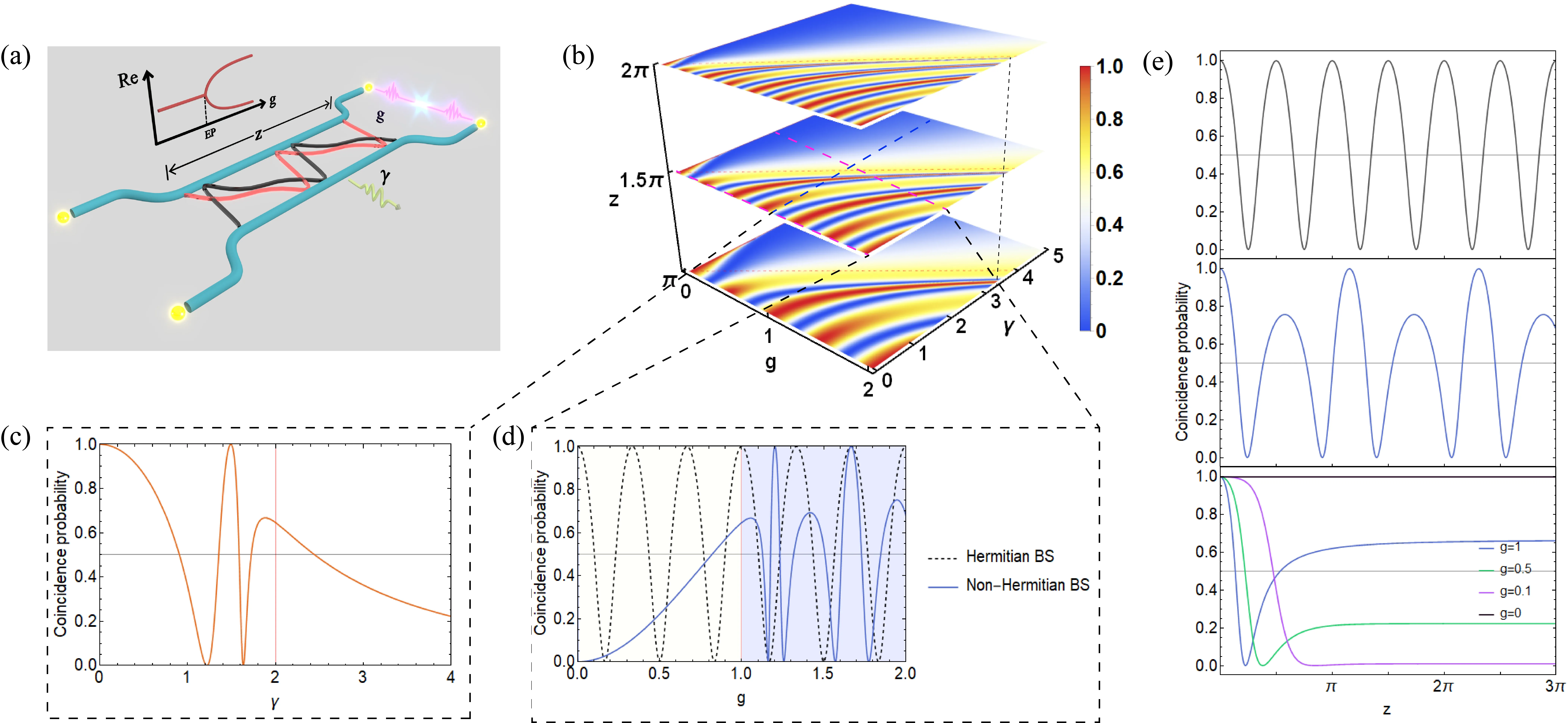
Exceptional-Point-Induced Sensitivity-Robustness Phase Transition in Quantum Interference
Quantum interference underpins many quantum information protocols but is typically studied in lossless Hermitian systems. Here, we reveal an exceptional point induced phase transition in two photon Hong Ou Mandel interference within a lossy coupled waveguide system. In the PT symmetric phase. interference is ultrasensitive to coupling strength, yielding sharp bunching antibunching switches. In the PT broken phase. it becomes robust oscillation free and propagation independent with coincidence probability stably tunable via coupling. These regimes enable enhanced quantum sensing and reliable two photon control for robust quantum information processing.
2511.16381Nov 2025
View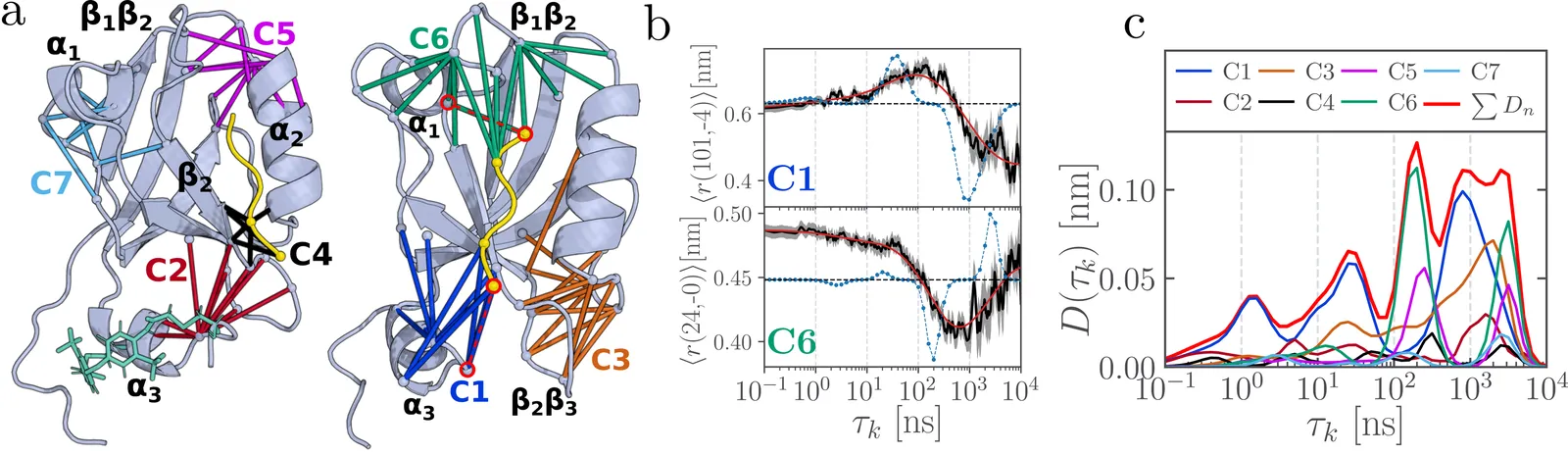
Contact cluster modeling of allosteric communication in PDZ domains
Allostery, the intriguing phenomenon of long-range communication between distant sites in proteins, plays a central role in biomolecular regulation and signal transduction. While it is commonly attributed to conformational rearrangements, the underlying dynamical mechanisms remain poorly understood. The contact cluster model of allostery [J. Chem. Theory Comput. 2024, 20, 10731] identifies localized groups of highly correlated contacts that mediate interactions between secondary structure elements. This framework proposes that allostery proceeds through a multistep process involving cooperative contact changes within clusters and communication between distant clusters, transmitted through rigid secondary structures. To demonstrate the validity and generality of the model, this Perspective employs extensive molecular dynamics simulations ($\sim 1\,$ms total simulation time) of four different photoswitchable PDZ domains and studies how different domains, ligands and perturbations influence both the contact clusters and their dynamical evolution. These analyses reveal several recurring clusters that represent shared flexible structural modules, such as loops connecting $β$-sheets, and show that the characteristic timescales of the nonequilibrium protein response can be directly associated with the motions of individual contact clusters. Thus, the dynamic decomposition of PDZ domains into contact clusters uncovers a modular, dynamics-based architecture that underlies and facilitates long-range allosteric communication.
2511.16355Nov 2025
View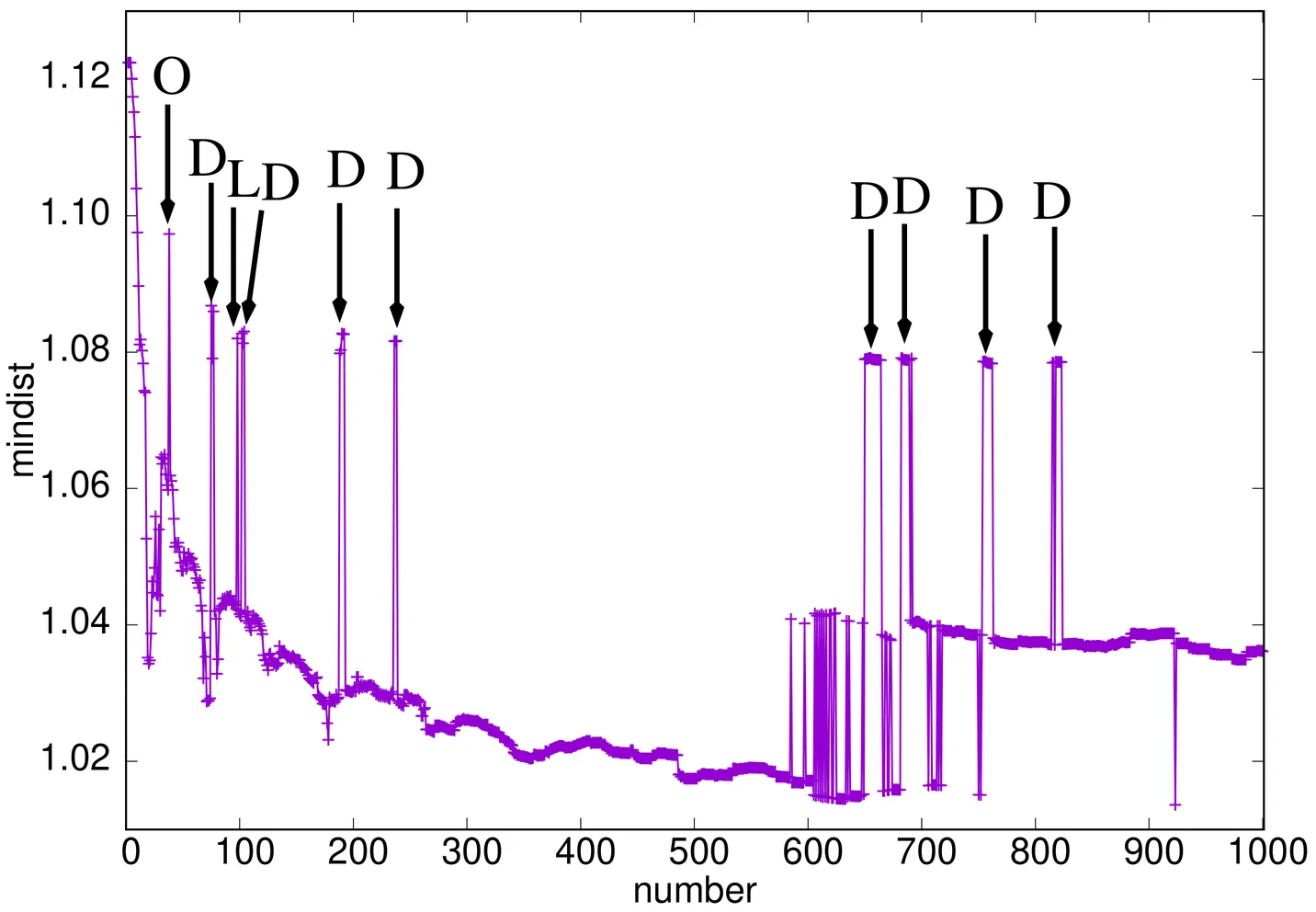
A note on the minimal pairwise distance in optimal Lennard-Jones $N$-body clusters
Good a-priori bounds on the smallest pairwise distance $r_{\rm{min}}(\mbox{LJ}_N^{\rm{gmin}})$ for a three-dimensional (3D) Lennard-Jones $N$-body cluster of globally minimal energy can significantly reduce the computational search space in the NP-hard problem to find this configuration. In this contribution the virial theorem is exploited for this purpose. We prove that if a configuration ${C}^{(N)}$ is a member of $\mbox{LJ}_N^{\rm{equ}}$ (the stationary points), then $r_{\rm{min}}({C}^{(N)}) \leq r_{\rm{min}}(\mbox{LJ}_2^{\rm{gmin}})$. It is also shown that if ${C}^{(N)}\in$ LJ$_N^{\rm{gmin}}\subset$ LJ$_N^{\rm{equ}}$, equality holds if and only if $N\in\{2,3,4\}$. We conjecture that $r_{\rm{min}}(\mbox{LJ}_N^{\rm{gmin}}) >1$ in units for which $r_{\rm{min}}(\mbox{LJ}_2^{\rm{gmin}})= 2^\frac16 \approx 1.122462048$. This conjectured lower bound, if correct, would improve the best lower bound currently known, $r_{\rm{min}}(\mbox{LJ}_N^{\rm{gmin}})\geq 0.767764$, by about 25$\%$. In these units the smallest minimal pair distance found through numerical searches for LJ$_N^{\rm{gmin}}$ with $N\leq 1000$ is $r_{\rm{min}}(\mbox{LJ}_{923}^{\rm{gmin}}) \approx 1.01361$, so the conjectured lower bound would presumably be close to optimal. From the virial theorem we obtain an identity for any ${C}^{(N)}\in \mbox{LJ}_N^{\rm{equ}}$, which expresses $r_{\rm{min}}({C}^{(N)})$ in terms of the distribution of relative distances in ${C}^{(N)}$. This result reveals interesting connections with the Erdős distance, and related problems.
2511.15008Nov 2025
View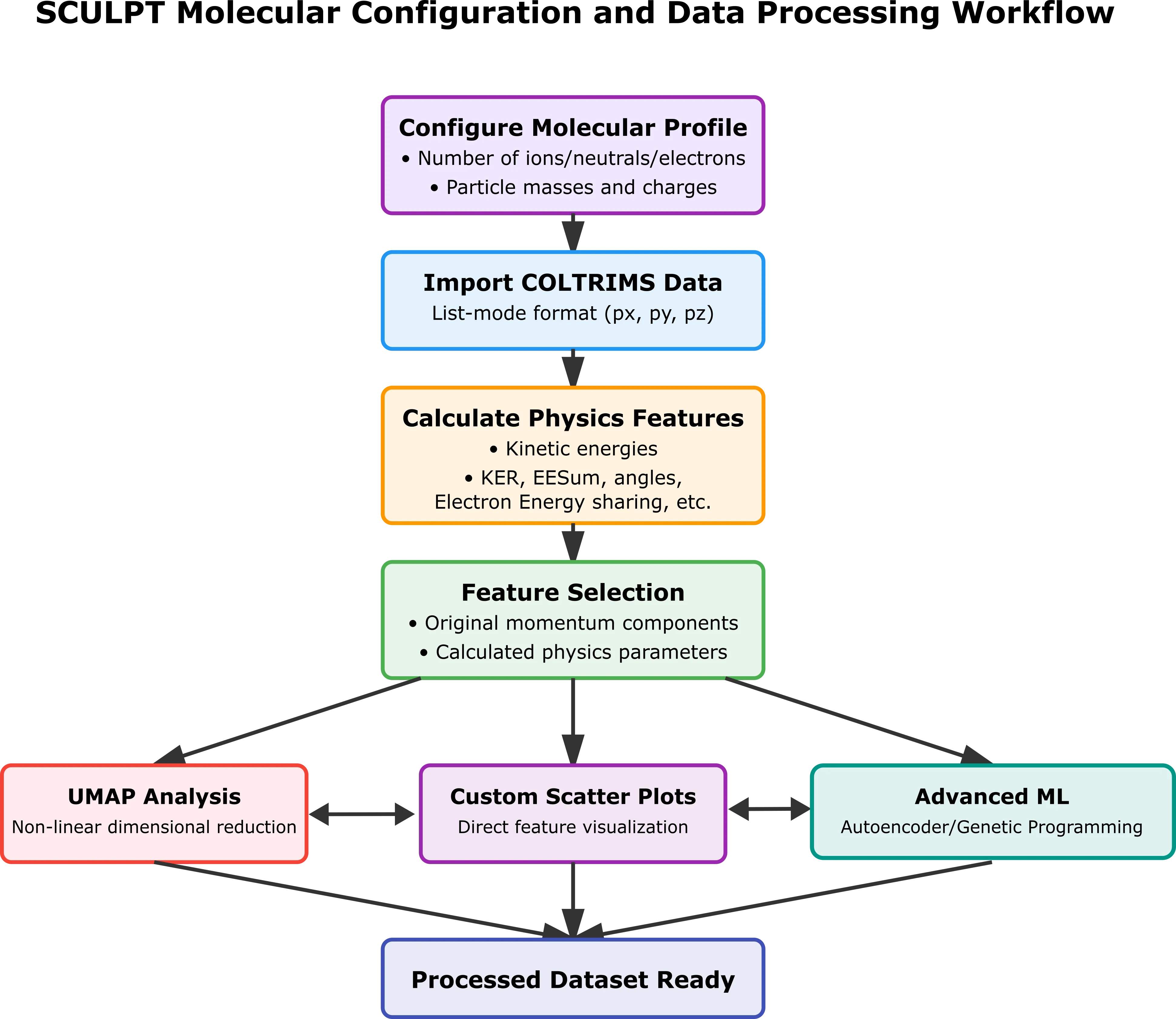
SCULPT: An Interactive Machine Learning Platform for Analyzing Multi-Particle Coincidence Data from Cold Target Recoil Ion Momentum Spectroscopy
We present SCULPT (Supervised Clustering and Uncovering Latent Patterns with Training), a comprehensive software platform for analyzing tabulated high-dimensional multi-particle coincidence data from Cold Target Recoil Ion Momentum Spectroscopy (COLTRIMS) experiments. The software addresses critical challenges in modern momentum spectroscopy by integrating advanced machine learning techniques with physics-informed analysis in an interactive web-based environment. SCULPT implements Uniform Manifold Approximation and Projection (UMAP) for non-linear dimensionality reduction to reveal correlations in highly dimensional data. We also discuss potential extensions to deep autoencoders for feature learning, and genetic programming for automated discovery of physically meaningful observables. A novel adaptive confidence scoring system provides quantitative reliability assessments by evaluating user-selected clustering quality metrics with predefined weights that reflect each metric's robustness. The platform features configurable molecular profiles for different experimental systems, interactive visualization with selection tools, and comprehensive data filtering capabilities. Utilizing a subset of SCULPT's capabilities, we analyze photo double ionization data measured using the COLTRIMS method for 3-body dissociation of the D2O molecule, revealing distinct fragmentation channels and their correlations with physics parameters. The software's modular architecture and web-based implementation make it accessible to the broader atomic and molecular physics community, significantly reducing the time required for complex multi-dimensional analyses. This opens the door to finding and isolating rare events exhibiting non-linear correlations on the fly during experimental measurements, which can help steer exploration and improve the efficiency of experiments.
2511.11153Nov 2025
View
Electron scattering from aminoacetonitrile: effects of polarisation-correlation and basis-set on cross section
Aminoacetonitrile occupies a prime importance in the interface between astrochemistry and prebiotic chemistry. Its detection in the ISM establishes it as part of the organic inventory of star-forming regions, while its role as a glycine precursor highlights its significance for origins-of-life scenarios. In this work, electron scattering from aminoacetonitrile has been studied using the $R$-matrix method in the low-energy range from $\sim$0 to 10 eV. The calculations were carried out within the $C_{s}$ point group using static-exchange (SE), static-exchange plus polarisation (SEP), and configuration interaction (CI) models, with two basis sets (6-311G* and cc-pVTZ) to understand their dependence on cross section. Various scattering observables, such as differential elastic cross section, integral elastic, excitation, and momentum transfer cross sections, were examined. Since aminoacetonitrile is a prebiotically relevant molecule, these findings provide valuable insight into electron-driven processes in complex organic systems and form a theoretical foundation for future work on electron-induced reactivity in prebiotic and astrophysical environments.
2509.18669Sep 2025
View
Electron Impact Fragmentation Dynamics of Carbonyl Sulfide: A Combined Experimental and Theoretical Study
In this study, we examine the interactions of low- to intermediate-energy electrons (0$-$45 eV) with carbonyl sulfide (OCS). These collisions lead to the formation of several anionic fragments, including C$^-$, O$^-$, S$^-$, and SO$^-$. When the incident electron energy is below the first ionization potential of the molecule, dissociative electron attachment (DEA) process dominates, primarily yielding O$^-$ and S$^-$ fragments. At higher energies, beyond the ionization potential, ion-pair dissociation (IPD) becomes the dominant process, resulting in the emergence of additional fragments such as C$^-$ and SO$^-$. This leads to an increasingly intricate mechanism, necessitating a detailed analysis to elucidate the ion-pair dissociation pathways. The absolute cross section for S$^-$ ions has been determined using the well-established relative flow technique. Theoretical cross sections are calculated using the multi-configurational time-dependent hartree (MCTDH) method, with each potential energy curve obtained from equation-of-motion coupled-cluster singles and doubles (EOM-CCSD) calculations. The computed values are in excellent agreement with the experimental data. The analysis reveals contributions from both linear and bent anionic resonant states. Due to low count rates, only relative cross section curves have been obtained for the O$^-$ and SO$^-$ ions. At higher energies, the ion pair thresholds are evaluated using the Wannier threshold law, yielding values consistent with those derived from thermochemical data.
2506.12528Jun 2025
View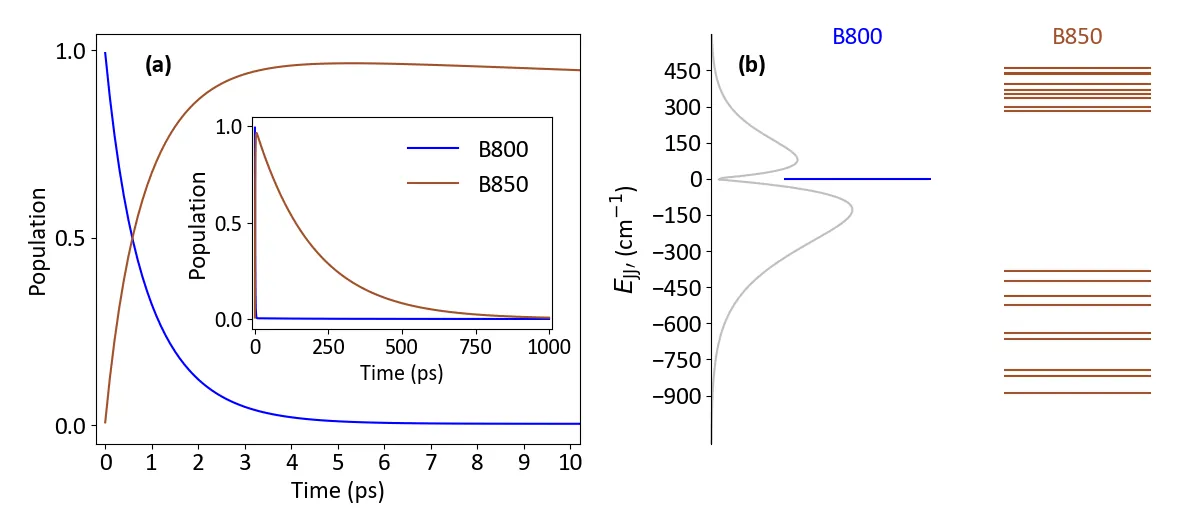
Microcavity-Enhanced Exciton Dynamics in Light-Harvesting Complexes: Insights from Redfield Theory
We investigated the exciton transfer dynamics in photosynthetic light-harvesting complex 2 (LH2) coupled to an optical microcavity. Using computational simulations based on Redfield theory, we analyzed how microcavity coupling influences energy relaxation and transfer within and between LH2 aggregates. Our results show that the exciton transfer rate between B850 rings follows a square dependence on the light-matter coupling strength, in agreement with Fermi's golden rule. Interestingly, the energy transfer rate remains almost independent of the number of LH2 complexes. This behavior is explained by the molecular components of the polaritonic wavefunction overlaps. These findings highlight the crucial role of cavity-induced polaritonic states in mediating energy transport and provide a theoretical framework for optimizing microcavity environments to enhance exciton mobility in light-harvesting systems and related photonic applications.
2504.02066Apr 2025
View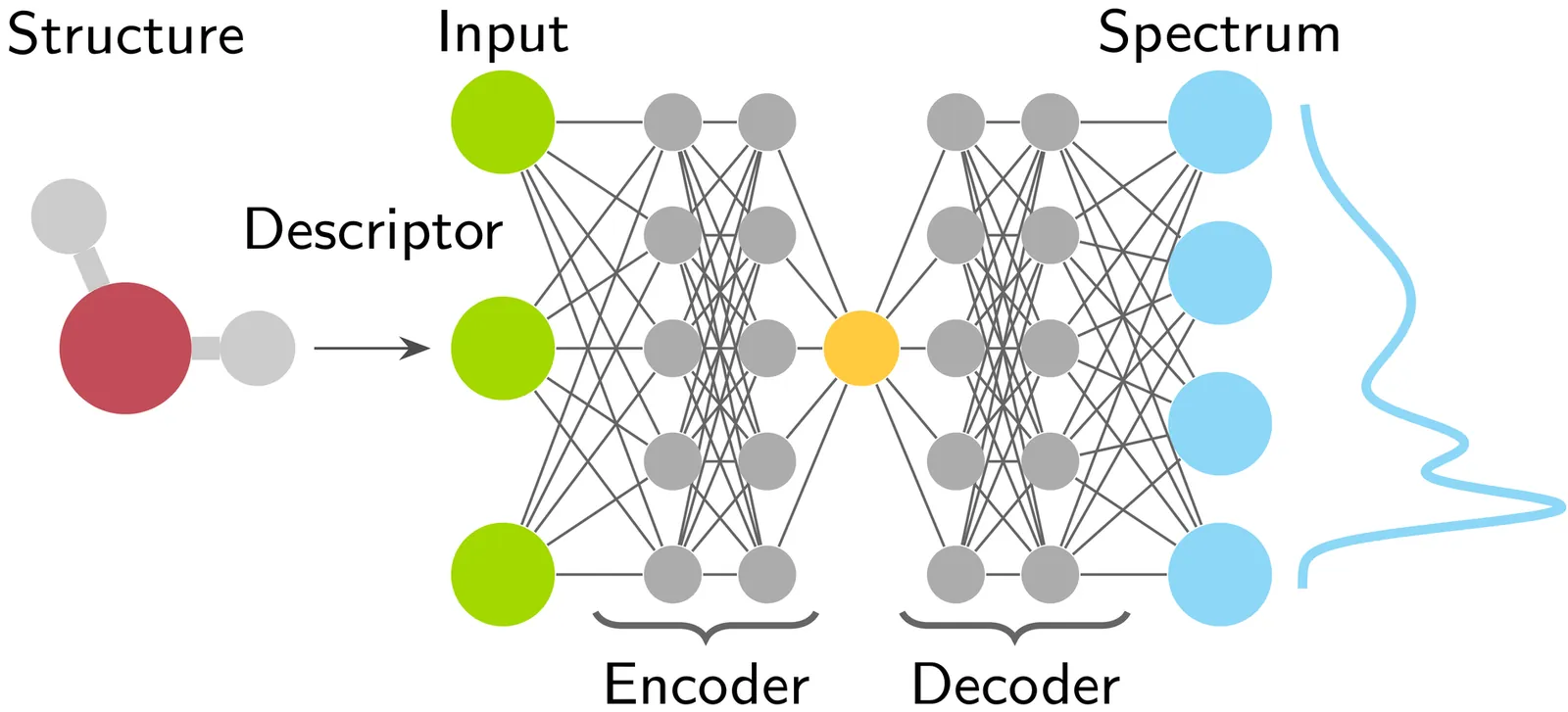
Encoder-Decoder Neural Networks in Interpretation of X-ray Spectra
Encoder--decoder neural networks (EDNN) condense information most relevant to the output of the feedforward network to activation values at a bottleneck layer. We study the use of this architecture in emulation and interpretation of simulated X-ray spectroscopic data with the aim to identify key structural characteristics for the spectra, previously studied using emulator-based component analysis (ECA). We find an EDNN to outperform ECA in covered target variable variance, but also discover complications in interpreting the latent variables in physical terms. As a compromise of the benefits of these two approaches, we develop a network where the linear projection of ECA is used, thus maintaining the beneficial characteristics of vector expansion from the latent variables for their interpretation. These results underline the necessity of information recovery after its condensation and identification of decisive structural degrees of freedom for the output spectra for a justified interpretation.
2406.140441Jun 2024
View