Trending in Atomic & Molecular Physics
AceFF: A State-of-the-Art Machine Learning Potential for Small Molecules
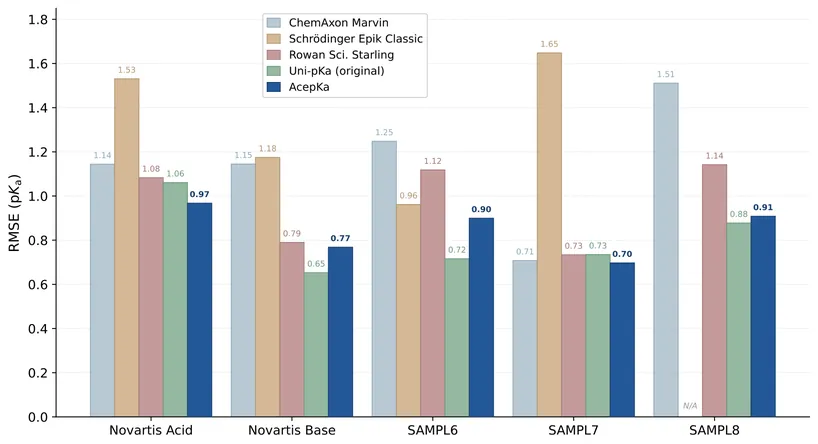
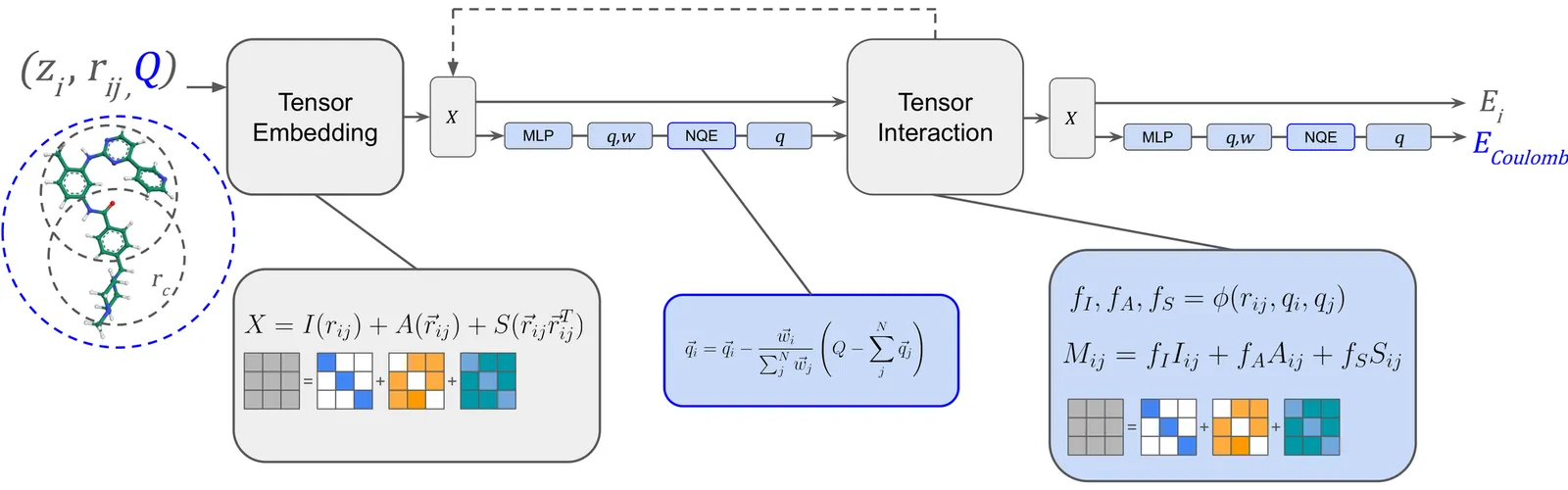
We introduce AceFF, a pre-trained machine learning interatomic potential (MLIP) optimized for small molecule drug discovery. While MLIPs have emerged as efficient alternatives to Density Functional Theory (DFT), generalizability across diverse chemical spaces remains difficult. AceFF addresses this via a refined TensorNet2 architecture trained on a comprehensive dataset of drug-like compounds. This approach yields a force field that balances high-throughput inference speed with DFT-level accuracy. AceFF fully supports the essential medicinal chemistry elements (H, B, C, N, O, F, Si, P, S, Cl, Br, I) and is explicitly trained to handle charged states. Validation against rigorous benchmarks, including complex torsional energy scans, molecular dynamics trajectories, batched minimizations, and forces and anergy accuracy demonstrates that AceFF establishes a new state-of-the-art for organic molecules. The AceFF-2 model weights and inference code are available at https://huggingface.co/Acellera/AceFF-2.0.
2601.00581
Jan 2026Chemical Physics