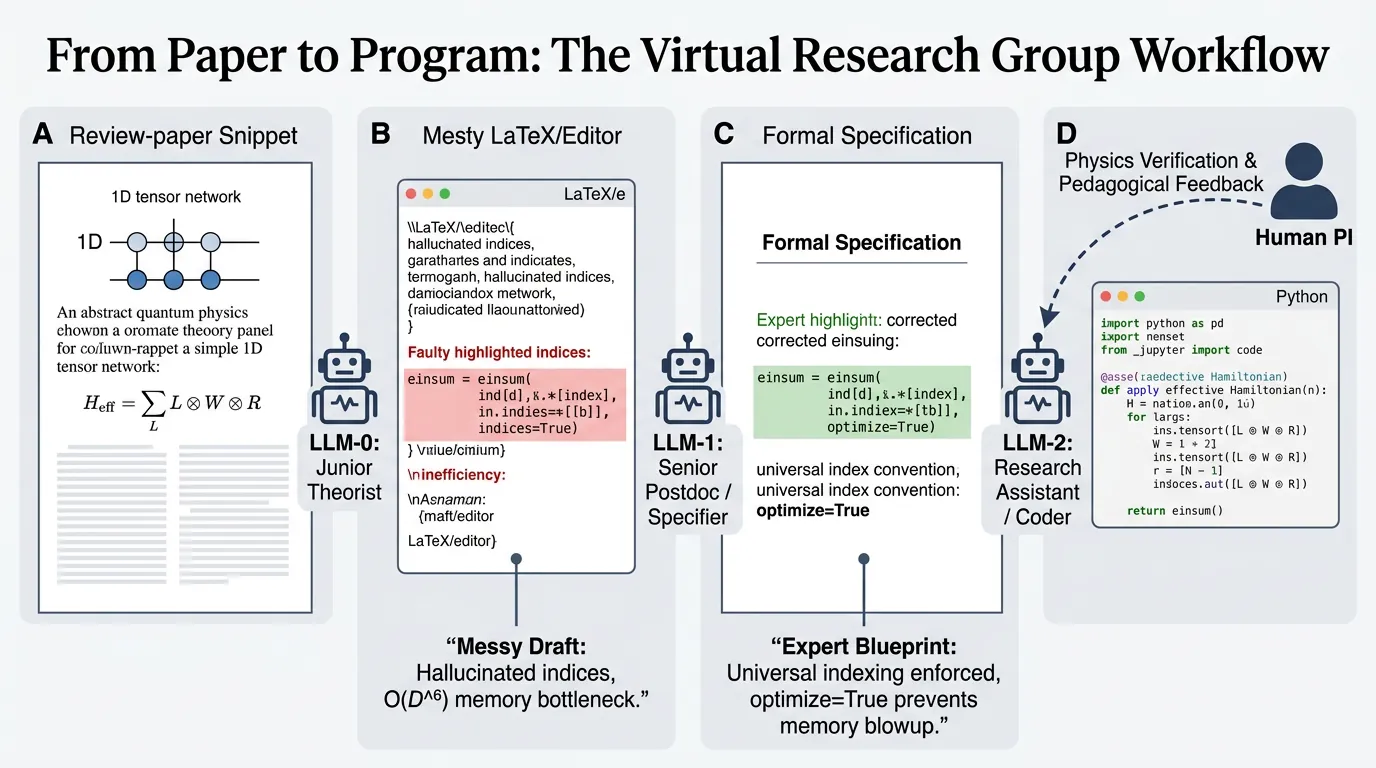
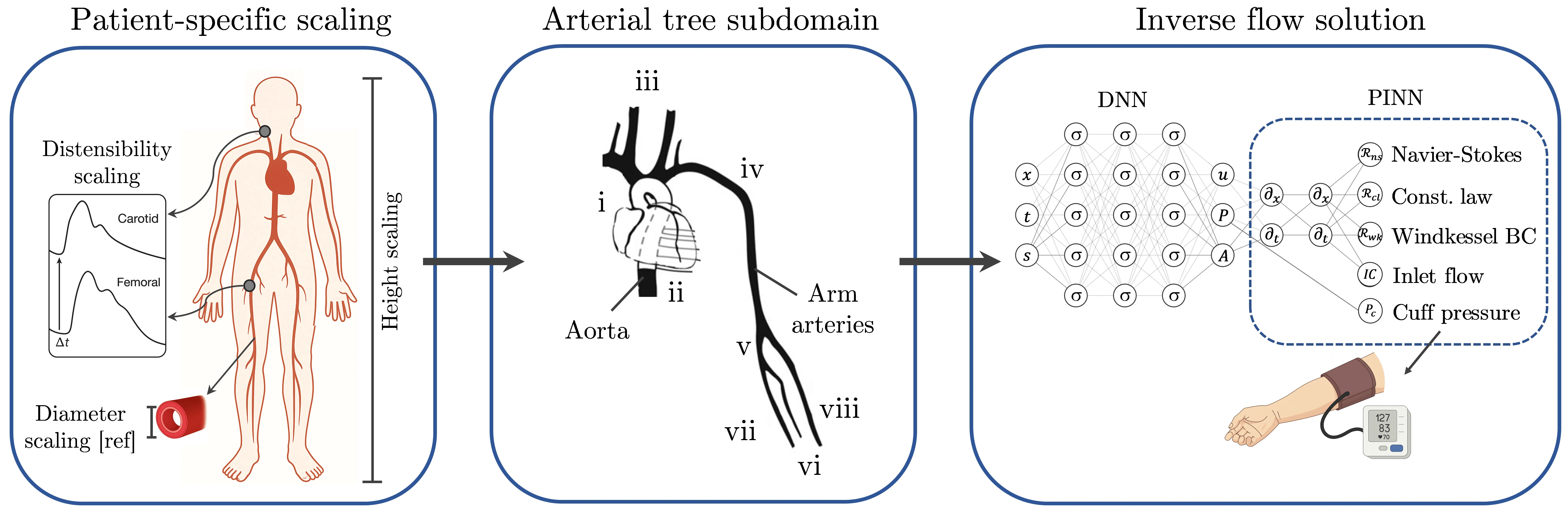
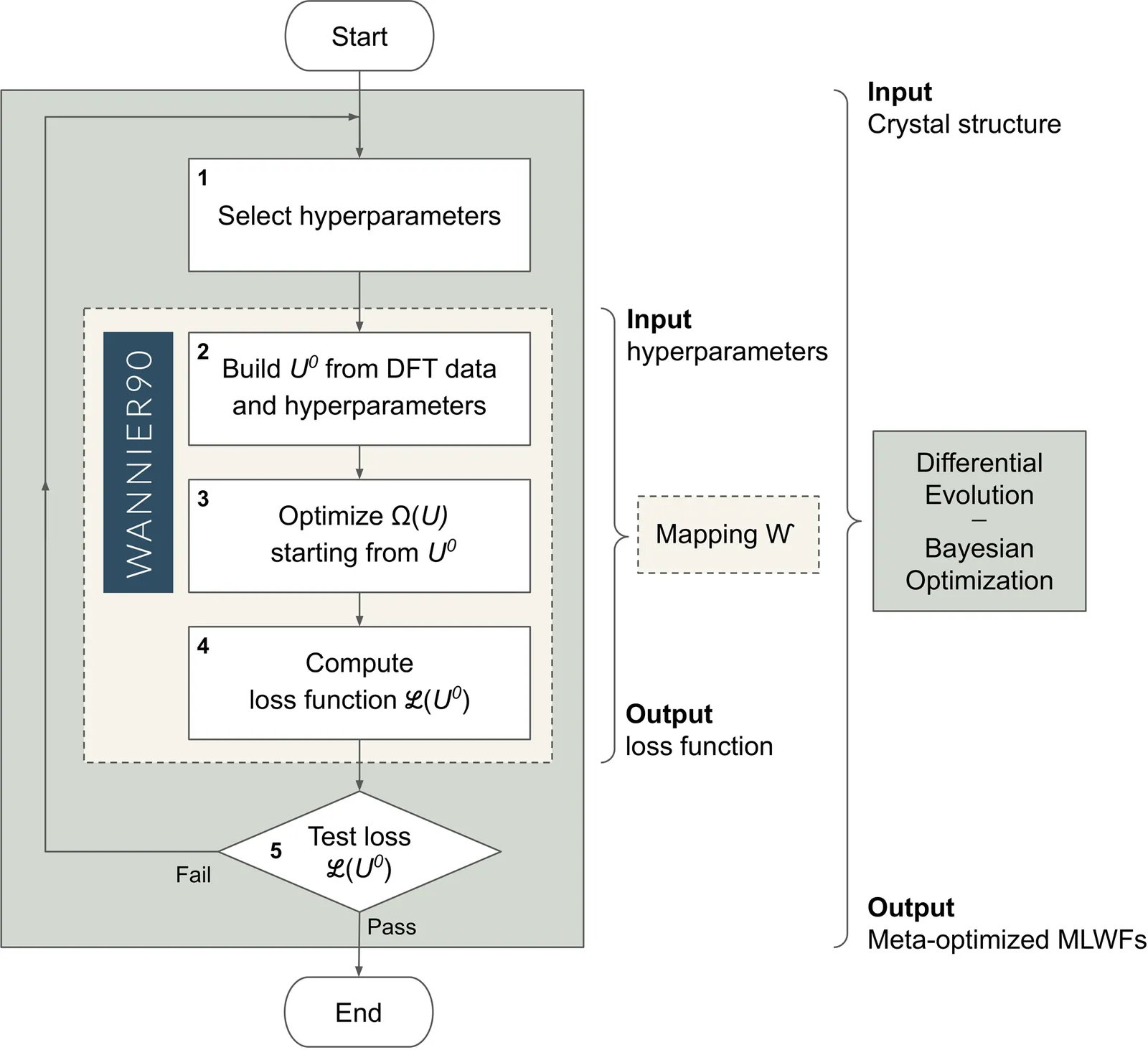
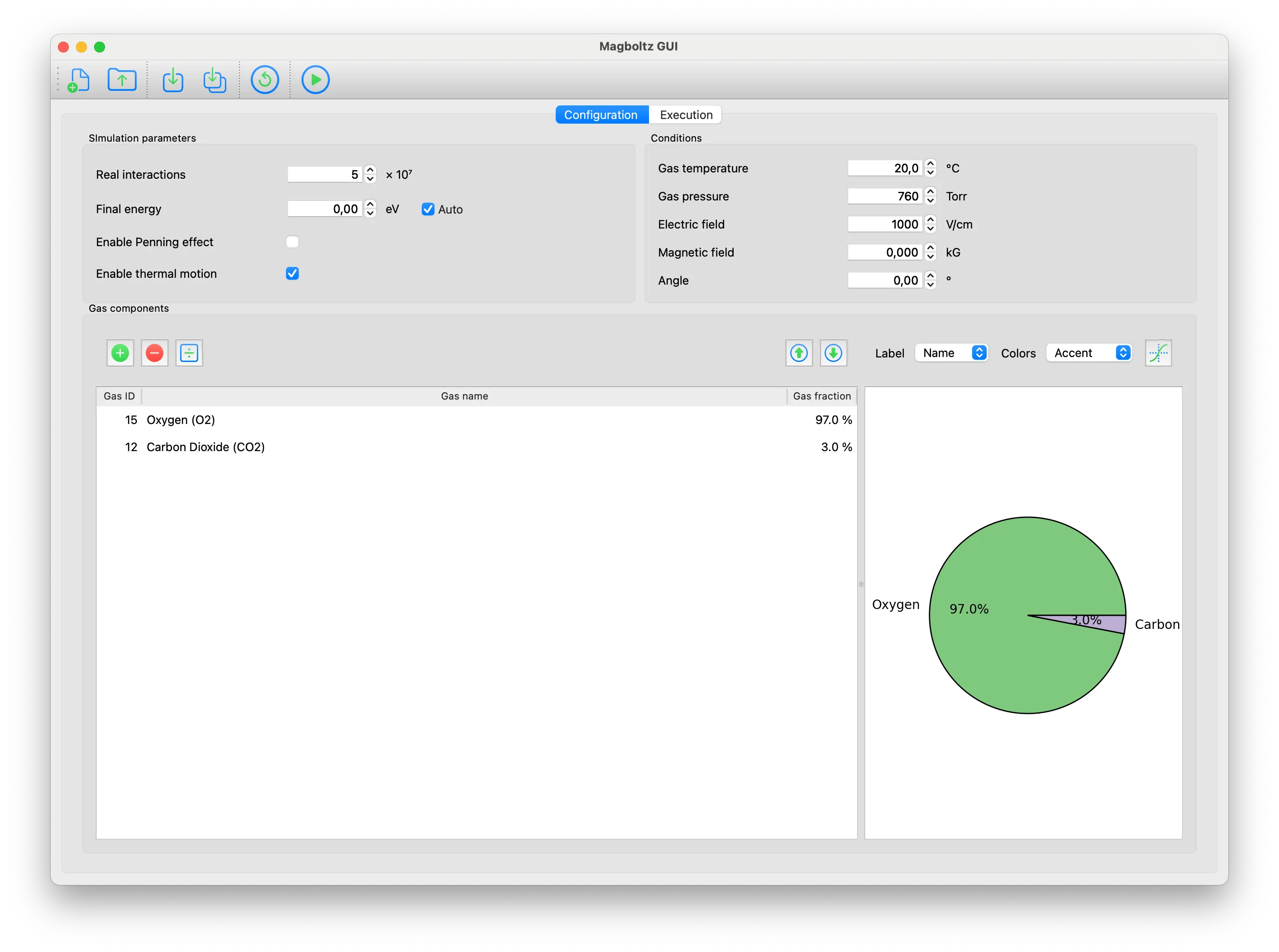
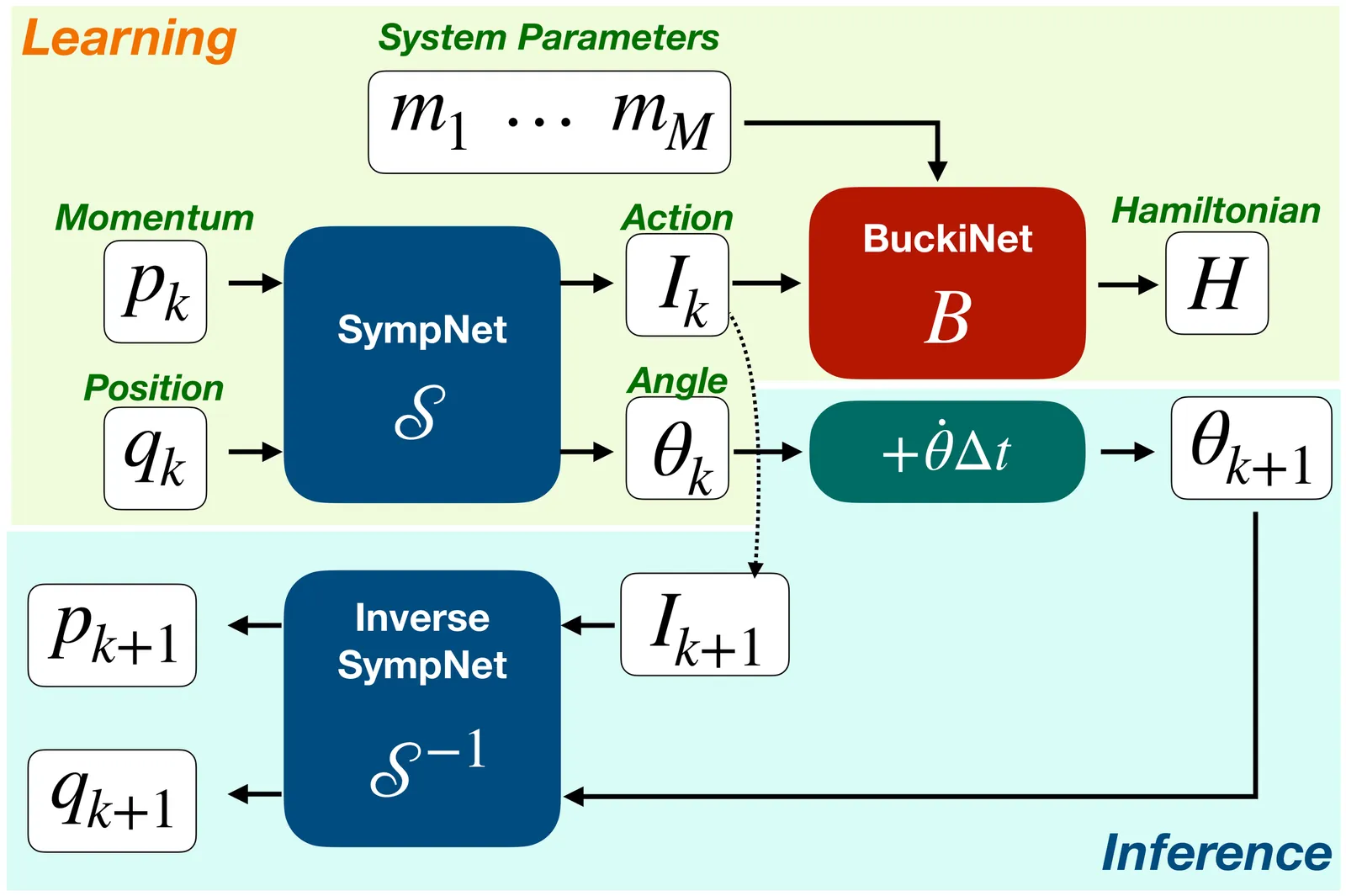
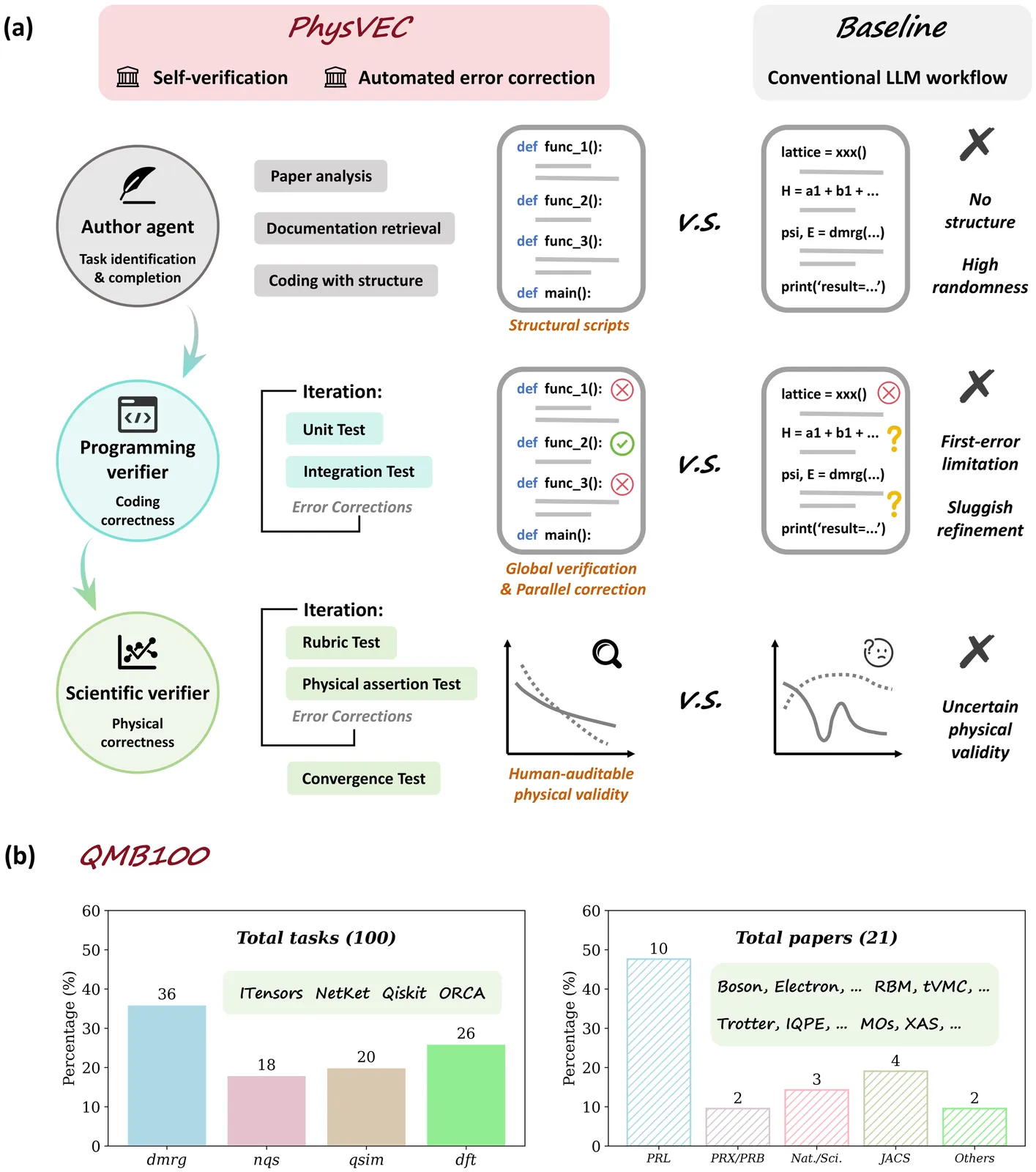
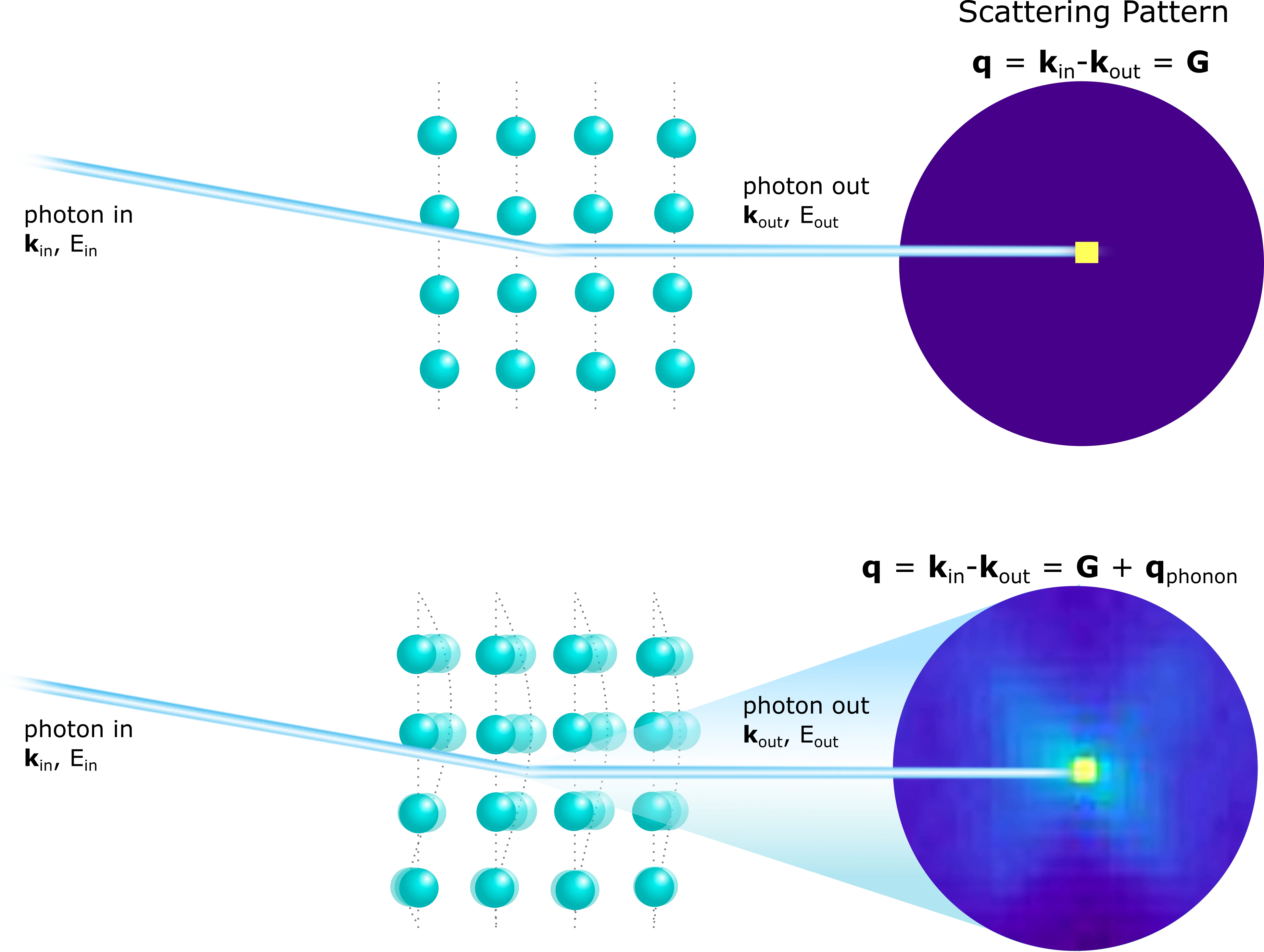
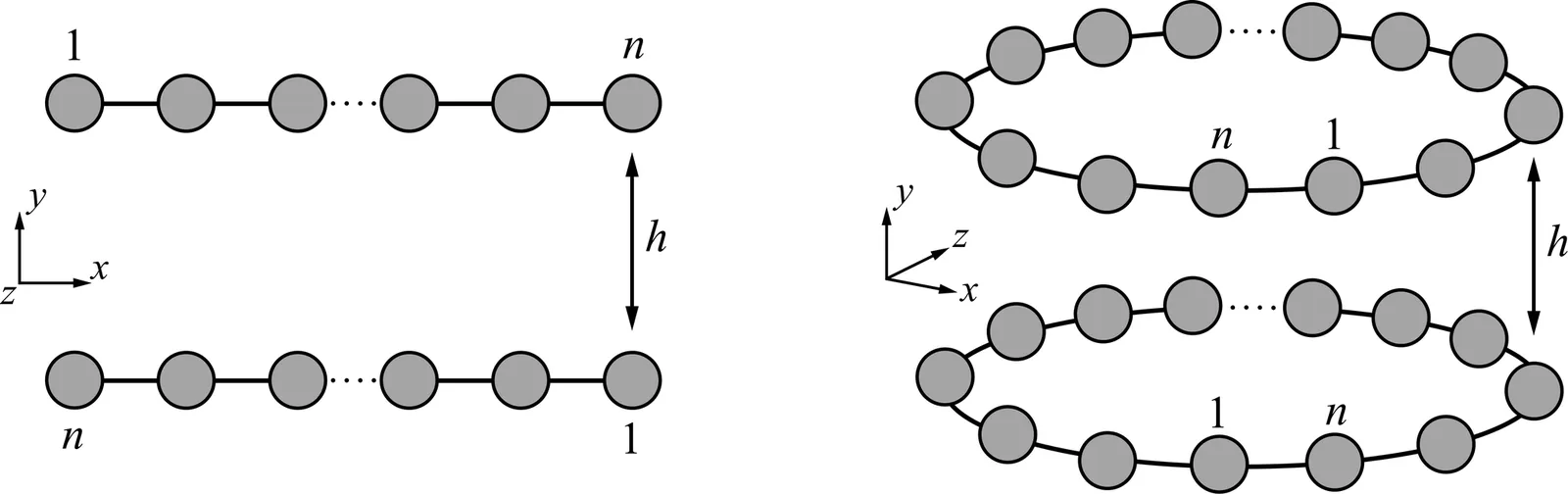
Trending in Computational Physics
qNEP: A highly efficient neuroevolution potential with dynamic charges for large-scale atomistic simulations
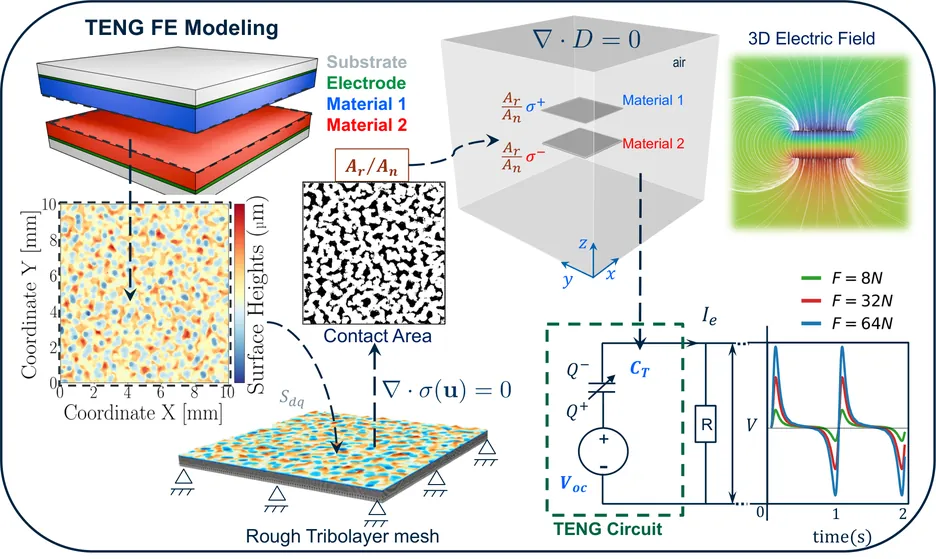
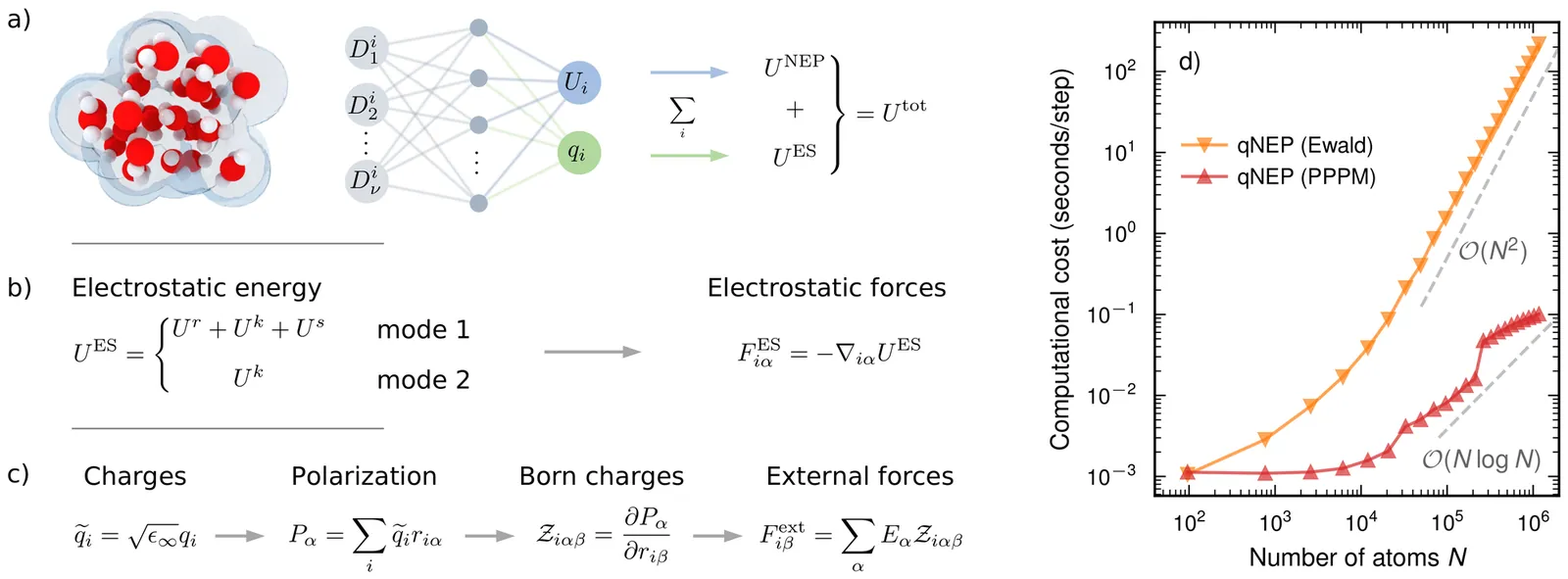
Although electrostatics can be incorporated into machine-learned interatomic potentials, existing approaches are computationally very demanding, limiting large-scale, long-time simulations of electrostatics-driven phenomena such as dielectric response, infrared activity, and field-matter coupling. Here, we extend the neuroevolution potential (NEP), a highly efficient machine-learned interatomic potential, to a charge-aware framework (qNEP) by introducing explicit, environment-dependent partial charges. Each ionic partial charge is represented by a neural network as a function of the local descriptor vector, analogous to the NEP site-energy model. This formulation enables the direct prediction of the Born effective charge tensor for each ion and, consequently, the polarization. As a result, dielectric properties, infrared spectra, and coupling to external electric fields can be evaluated within a unified framework. We derive consistent expressions for the forces and virials that explicitly account for the position dependence of the partial charges. The qNEP method has been implemented in the free-and-open-source GPUMD package, with support for both Ewald summation and particle-particle particle-mesh treatments of electrostatics. We demonstrate the accuracy and efficiency of the qNEP approach through representative applications to water, Li7La3Zr2O12, BaTiO3, and a magnesium-water interface. These results show that qNEP enables accurate atomistic simulations with explicit long-range electrostatics, scalable to million-atom systems on nanosecond time scales using consumer-grade GPUs.
2601.19034
Jan 2026Computational Physics
Stable spectral neural operator for learning stiff PDE systems from limited data
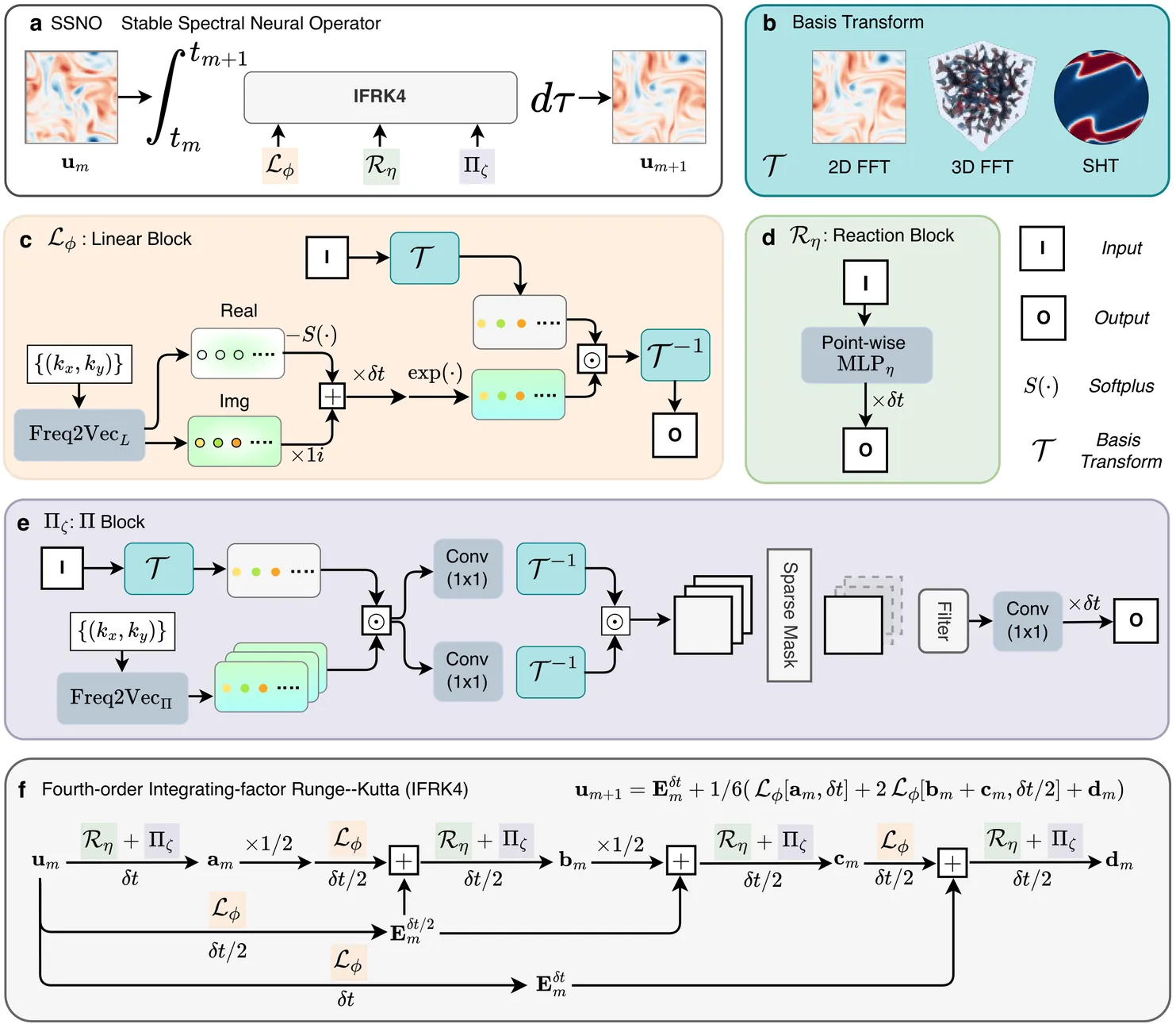
Accurate modeling of spatiotemporal dynamics is crucial to understanding complex phenomena across science and engineering. However, this task faces a fundamental challenge when the governing equations are unknown and observational data are sparse. System stiffness, the coupling of multiple time-scales, further exacerbates this problem and hinders long-term prediction. Existing methods fall short: purely data-driven methods demand massive datasets, whereas physics-aware approaches are constrained by their reliance on known equations and fine-grained time steps. To overcome these limitations, we introduce an equation-free learning framework, namely, the Stable Spectral Neural Operator (SSNO), for modeling stiff partial differential equation (PDE) systems based on limited data. Instead of encoding specific equation terms, SSNO embeds spectrally inspired structures in its architecture, yielding strong inductive biases for learning the underlying physics. It automatically learns local and global spatial interactions in the frequency domain, while handling system stiffness with a robust integrating factor time-stepping scheme. Demonstrated across multiple 2D and 3D benchmarks in Cartesian and spherical geometries, SSNO achieves prediction errors one to two orders of magnitude lower than leading models. Crucially, it shows remarkable data efficiency, requiring only very few (2--5) training trajectories for robust generalization to out-of-distribution conditions. This work offers a robust and generalizable approach to learning stiff spatiotemporal dynamics from limited data without explicit \textit{a priori} knowledge of PDE terms.
2512.11686
Dec 2025Computational Physics